Metirapona ▼ Metopirone® (HRA Pharma) en diagnóstico de la insuficiencia de ACTH y síndrome de Cushing
 Nº394
Nº394

Resumen
La metirapona es un inhibidor de la síntesis de aldosterona y cortisol en la corteza suprarrenal, bloqueando de forma reversible y selectiva la 11β-hidroxilasa y, adicionalmente, la 18-hidroxilasa. El resultado es una reducción de los niveles de cortisol y, especialmente, de aldosterona. El medicamento ha sido autorizado como prueba diagnóstica para la insuficiencia de ACTH y en el diagnóstico diferencial del síndrome de Cushing ACTH-dependiente, así como para el manejo de pacientes con síndrome de Cushing endógeno. En general, se considera que la prueba de la metirapona en el diagnóstico del síndrome de Cushing tiene una sensibilidad y especificidad muy similar a la prueba de supresión con altas dosis de dexametasona, con la ventaja de que la combinación de los resultados de ambas pruebas es aún más fiable que cada una de ellas por separado. Por lo que respecta a su uso terapéutico en el síndrome de Cushing, los datos son lo suficientemente claros como para garantizar su utilidad, al menos en una proporción relevante de pacientes. Actualmente, la única alternativa farmacológica a la metirapona en el tratamiento del síndrome de Cushing es el ketoconazol, con efectos equiparables. Por ello, la decisión de usar uno u otro, debe ser adoptada de forma personalizada, teniendo en cuenta sobre todo los potenciales efectos adversos del tratamiento. Ambas opciones, son aceptables en pacientes donde otras opciones, como la cirugía o la radioterapia, no sean aconsejables o no hayan producido los resultados buscados.
ASPECTOS FISIOPATOLÓGICOS
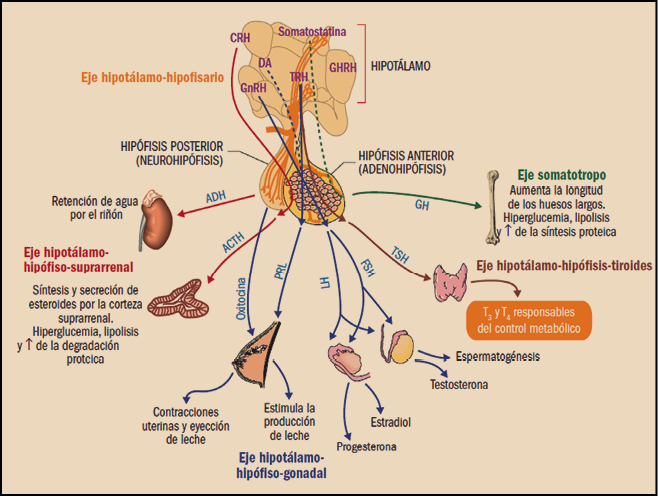
La hipófisis es una glándula del tamaño de un guisante con un diámetro aproximado de 1,3 cm, localizada en la silla turca del hueso esfenoidal y, por lo tanto, protegida en toda su superficie salvo por la cara superior, por la que se conecta con el hipotálamo a través de una estructura denominada infundibulum o tallo hipotálamo-hipofisario. Por consiguiente, está estrechamente relacionada con el hipotálamo, principal centro integrador común de control de la función de los órganos endocrinos, mediante vías neuronales y hormonales. De hecho, influido por impulsos recibidos del sistema nervioso central (SNC), controla y modula el funcionamiento de la hipófisis, merced a su íntima relación tanto anatómica como funcional. A su vez, las hormonas hipofisarias modulan a los centros hipotalámicos mediante un retrocontrol o feedback corto. Tal es la integración de ambos que al conjunto se lo denomina sistema hipotálamo-hipofisario (Tresguerres, 2008).
La hipófisis tiene dos partes anatómica y funcionalmente distintas: la anterior (adenohipófisis) y la posterior (neurohipófisis). La adenohipófisis supone el 75% del peso total, sus células son de tipo epitelial glandular y secretan varias hormonas; por parte, la neurohipófisis contiene axones y terminales axónicas de cerca de 5.000 neuronas, cuyos cuerpos neuronales se encuentran en los núcleos paraventricular y supraóptico del hipotálamo. En realidad, es en los cuerpos neuronales situados en el hipotálamo donde se generan las hormonas de la hipófisis posterior (oxitocina y vasopresina), de forma que ésta actúa como un mero reservorio de hormonas. Los axones atraviesan el tallo hipotálamo-hipofisario, generando una conexión directa entre el hipotálamo y la neurohipófisis. Además de los axones antes mencionados, existen también algunas escasas células de sostén denominadas pituicitos. Existe una tercera región hipofisaria denominada lóbulo intermedio, que se atrofia durante el desarrollo fetal y es muy pequeño en los adultos, inmigrando sus células a la adenohipófisis.
Durante mucho tiempo, la hipófisis ha sido denominada glándula directora del concierto endocrino, pues sus hormonas controlan a la mayoría de las otras glándulas endocrinas y a otros órganos. En realidad, dicho título debería ser atribuido al hipotálamo, pues es éste el que controla las dos partes de la hipófisis; de hecho, existen hormonas de la hipófisis anterior que están bajo el control de hormonas estimuladoras hipotalámicas, y otras que están bajo el control de hormonas fundamentalmente inhibidoras, como es el caso de la prolactina. Existe finalmente un tercer tipo de hormonas que están sometidas a un control doble por parte de hormonas estimulantes e inhibidoras, cuyo ejemplo fundamental es precisamente la hormona de crecimiento.
La adenohipófisis secreta seis hormonas peptídicas importantes1 que se clasifican, según su estructura, en tres grupos:
- Hormonas somatotrópicas: hormona de crecimiento (GH) y prolactina (PRL).
- Hormonas glucoproteicas: tirotropina (TSH), hormona luteinizante (LH) y hormona foliculoestimulante (FSH).
- Hormonas derivadas de la proopiomelanocortina (POMC), cuyo principal exponente es la hormona adrenocorticotropa o corticotropina (ACTH).
Por su parte, las principales hormonas hipotalámicas estimulantes de la adenohipófisis son la hormona liberadora de la hormona de crecimiento (somatorelina, somatoliberina o GHRH), la hormona liberadora de las gonadotropinas (gonadorelina o GnRH; también conocida como hormona liberadora de hormona luteinizante, LHRH), la hormona liberadora de tirotropina (tirorelina o TRH) y la hormona liberadora de la corticotropina (corticorelina, corticoliberina o CRH). Por su parte, las hormonas hipotalámicas inhibidoras incluyen la somatostatina, que inhibe la liberación de hormona de crecimiento, y la dopamina, que inhibe la secreción de prolactina.
En el ámbito del eje hipotálamo hipofiso-suprrenal, la hiperfunción de las glándulas suprarrenales puede producir un aumento de la secreción de todas las hormonas generadas en la corteza de dichas glándulas. Desde un punto de vista clínico, las enfermedades más importantes son las debidas al aumento de la secreción de glucocorticoides, globalmente denominadas como síndrome de Cushing. Las causas pueden ser una secreción incrementada de corticotropina (ACTH) por la hipófisis o un tumor extrahipofisario (secreción ectópica de ACTH), o bien un aumento excesivo de la secreción de cortisol por la corteza suprarrenal. Por su parte, la hiperprodución de mineralcorticoides o hiperaldosteronismo puede ser primaria, debida a un adenoma suprarrenal, o bien secundaria, debida a la activación del sistema renina-angiotensina. Finalmente, merece la pena citar al feocromocitoma, un tumor de la médula suprarrenal.
La causa más común del síndrome de Cushing es la iatrogénica, producida por el tratamiento prolongado con prednisona u otros glucocorticoides. Entre los síndromes de Cushing espontáneos, los más frecuentes se deben a tumores hipofisarios hiperproductores de ACTH y, en ocasiones, a alteraciones del sistema nervioso que secrete CRH (hormona liberadora de corticotropina, o corticoliberina) en exceso; esto es lo que se denomina enfermedad de Cushing. Los tumores suprarrenales, que secretan cortisol de manera autónoma, pueden ser también parte de los síndromes de Cushing.
La enfermedad de Cushing tiene mayor incidencia en mujeres que en varones, siendo la proporción entre ambos sexos de aproximadamente 8:1. En estos casos aumenta también la secreción de andrógenos por las suprarrenales (DHEA, DHEA sulfato y androstenediona), mientras que la secreción de aldosterona no se incrementa. Este aumento de los andrógenos suprarrenales en las mujeres con enfermedad de Cushing puede producir hirsutismo, acné y amenorrea. En los varones, el exceso de cortisol inhibe la esteroidogénesis testicular, lo que puede producir disminución de la libido e impotencia.
La obesidad es el síntoma más frecuente (más del 90% de los casos) del síndrome de Cushing. Afecta fundamentalmente al tronco, mientras que, por el contrario, disminuye el depósito de grasa en las extremidades. También se acumula grasa en el cuello (cuello de búfalo) y la cara se redondea, proporcionándole aspecto de media luna. Los glucocorticoides en exceso tienen efectos catabólicos, con destrucción de proteínas en la mayoría de los tejidos. Producen atrofia de la epidermis y estrías de color púrpura en la piel; los músculos se debilitan, hay pérdida de la masa y del tono muscular, sobre todo en las extremidades inferiores, lo que produce fatiga al realizar un esfuerzo. El sistema inmunitario se altera y aumenta la frecuencia de las infecciones oportunistas. El aumento de cortisol produce un incremento de la resorción ósea y una disminución de la formación del hueso, lo que da lugar en los adultos a osteoporosis y mayor frecuencia de fracturas óseas. En el 75% de los pacientes se produce hipertensión, uno de los factores que antiguamente contribuía a la elevada mortalidad de los enfermos sin tratar. En un tercio de los pacientes se producen alteraciones psicológicas.
Se estimada que el 2-3% de los pacientes con diabetes de difícil control y obesidad pueden tener un síndrome de Cushing no reconocido, es decir “hipercortisolismo subclínico” de ahí que se requiera de estudios altamente sensibles que permitan una adecuada discriminación bioquímica entre los pacientes con verdadero hipercortisolismo y aquellos con síndrome metabólico y fenotipo cushingoide. El proceso de identificación o localización de la causa del síndrome de Cushing una vez que se ha confirmado el hipercortisolismo diversas pruebas para poder diferenciar su origen: hipofisario, ectópico o suprarrenal. Desafortunadamente no existen pruebas que por sí solas puedan ser consideradas como la mejor en términos de efectividad y disponibilidad para el escrutinio y la localización del síndrome de Cushing y en ocasiones por lo tanto será necesaria más de una (Espinosa de los Monteros, 2005).
Una de las pruebas utilizadas en el escrutinio es la supresión con dosis bajas de dexametasona. Está basada en que, en condiciones normales, tras la administración de 1 mg de dexametasona a las 23.00 h, el cortisol plasmático a las 8.00 h del día siguiente debe ser inferior a 1,8 μg/dl. La sensibilidad alcanza el 93-96%. Sin embargo, hasta un 3% de los pacientes con síndrome de Cushing muestran supresión, en probable relación con la actividad cíclica, y en alguna serie se ha detectado hasta el 30% de falsos positivos en individuos sin síndrome de Cushing. Dosis mayores de 1 mg no mejoran la discriminación. La prueba de la dexametasona-CRH consiste en administrar de 2 mg/día de dexametasona durante 2 días, por vía oral, y aplicar a las 8.00, es decir 2 h después de la última dosis de dexametasona, una prueba de CRH (100 μg iv). Los pacientes con síndrome de Cushing responden a los 15 min de la inyección de CRH con un valor de cortisol en plasma por encima de 1,4 μg/dl, mientras que aquellos con seudo-Cushing muestran cifras inferiores. La prueba posee una capacidad discriminativa aceptable y carece de efectos secundarios. Los resultados iniciales mostraron sensibilidad y especificidad del 100%, aunque tanto la anorexia nerviosa como el ejercicio pueden generar respuestas similares al síndrome de Cushing.
Con la prueba de la desmopresina los pacientes con enfermedad de Cushing de origen hipofisario responden en el 80 al 90% de los casos a la administración de desmopresina (10 μg iv) con liberación de ACTH debido a la expresión de receptores V2 de la vasopresina en las células adenomatosas. Se considera respuesta positiva cuando el aumento de ACTH respecto al valor basal es > 6 pmol/l. No obstante, los resultados son variables pues se han observado respuestas positivas hasta en el 36% de los pacientes con depresión y en el 10% de los normales.
Aunque la loperamida inhibe la CRH y suprime los niveles de ACTH y de cortisol en individuos normales (seudo-Cushing) pero no en aquellos con síndrome de Cushing verdadero, la administración oral de 16 mg de loperamida no ha mostrado consistentemente superiores a las pruebas anteriores para diferenciar el síndrome de Cushing del seudo-Cushing. Tampoco con la prueba de inducción de hipoglucemia mediante la administración de insulina.
Por último, merece la pena citar entre las pruebas farmacológicas de interés en diagnóstico funcional de la enfermedad de Cushing a la prueba de supresión de dosis alta con dexametasona, que permite diferenciar el origen (hipofisario vs. ectópico) los cuadros dependientes de ACTH. Su fundamento es la capacidad de la dexametasona (administrada en dosis alta) para actuar sobre los receptores glucocorticoideos en el tumor hipofisario suprimiendo la producción de ACTH y por lo tanto de cortisol, mientras que en los casos de Cushing ectópico debido a la ausencia de estos receptores, no se presentará dicha supresión. En general, la sensibilidad de las pruebas de supresión con dosis altas de dexametasona oscila entre el 60 y el 80%, y la especificidad, entre el 60 y el 90% cuando se toma como punto de corte la reducción de cortisol plasmático del 50%. Hay tres estrategias para llevar a cabo esta prueba: 2 mg/6 h por vía oral durante 2 días, 8 mg por vía oral en dosis única nocturna a las 23.00 h o infusión intravenosa de 1 mg/h de dexametasona durante 5 o 7 h. No obstante, aproximadamente el 20-30% de los pacientes con adenomas hipofisarios no suprimen tras dexametasona oral, especialmente macroadenomas, y un porcentaje similar de ectópicos lo hacen, por lo que las pruebas de supresión con dexametasona no son definitivas (Cuéllar, 2014).
Actualmente, la cirugía constituye la única opción curativa, mediante la eliminación de la fuente del exceso de ACTH (corticotropina) o de cortisol. Sin embargo, esto no siempre es posible; de hecho, las tasa de remisión de la enfermedad de Cushing tras cirugía de la hipófisis oscilan entre un 40% y un 90% (Biller, 2008) y, además, implica un riesgo inaceptable para algunos pacientes con enfermedad de Cushing adrenal o extra-adrenal. Por este motivo, la utilización de fármacos capaces de inhibir determinados enzimas responsables de la esteroidogénesis sigue siendo, por el momento, la principal opción terapéutica.
Dos son los principales fármacos utilizados actualmente en la inhibición de la esteroidogénesis de la corteza adrenal, susceptibles de reducir de forma reversible la síntesis de cortisol; se trata del ketoconazol y de la metirapona; aunque el mitotano también es capaz de actuar a este nivel, su carácter de inhibidor irreversible le ha relegado al tratamiento sintomático del carcinoma avanzado de la corteza suprarrenal (adrenocortical) inextirpable, metastásico o recidivante (Daniel, 2015).
El ketoconazol es un conocido antifúngico imidazólico que, en dosis superiores a las utilizadas en micosis sistémicas (400-1600 vs. 200-400 mg/día), es capaz de reducir de forma significativa la producción adrenocortical de cortisol, debido a su acción sobre diversos enzimas implicados en la síntesis de aldosterona, cortisol y testosterona a partir de colesterol. En concreto, actúa de forma reversible sobre varios enzimas del citocromo P450: 17α-hidrolasa, 20,22-desmolasa, 11β-hidroxilasa, 17,20-desmolasa y 18-hidroxilasa. Igualmente, bloquea la síntesis de testosterona en las gónadas; de hecho, éste fue el principal motivo por el cual su uso como antifúngico fue suspendido en el año 2013 (AEMPS, 2013). El mayor estudio retrospectivo realizado con ketoconazol en síndrome de Cushing incluyó a 200 pacientes franceses (Castinetti, 2014), un 75% de los cuales respondió al tratamiento con dosis de 200 a 1.200 mg/día, normalizando los niveles urinarios de cortisol libre en el 49%.
El otro fármaco utilizado es el que ahora acaba de ser comercializado en España. Se trata de la metirapona, un inhibidor de la esteroidogénesis adrenocortical más selectivo que el ketoconazol y que afecta exclusivamente a la síntesis de aldosterona y de cortisol a este nivel, sin afectar significativamente a la de testosterona.
ACCIÓN Y MECANISMO
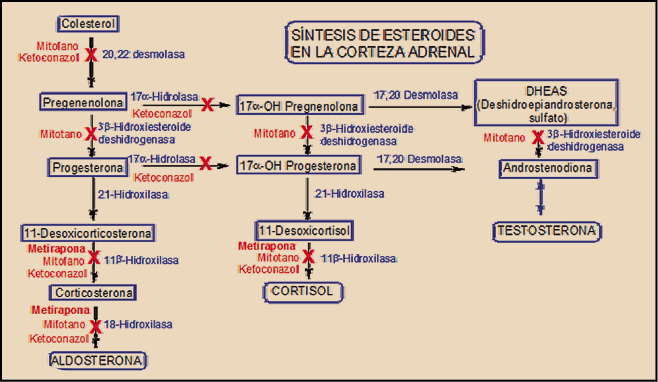
La metirapona es un inhibidor de la síntesis de aldosterona y cortisol en la corteza suprarrenal, bloqueando de forma reversible y selectiva la 11β-hidroxilasa y, adicionalmente, la 18-hidroxilasa. El resultado es una reducción de los niveles de cortisol y, especialmente, de aldosterona. El medicamento ha sido autorizado como prueba diagnóstica para la insuficiencia de ACTH y en el diagnóstico diferencial del síndrome de Cushing ACTH-dependiente, así como para el manejo de pacientes con síndrome de Cushing endógeno.
La reducción de los niveles de cortisol impide el efecto de retrocontrol inhibitorio del cortisol sobre la secreción adrenocrotical de sus precursores (11-desoxicorticosterona y 11-desoxicortisol), lo cual se traduce en un incremento de la producción de ACTH (corticotropina) por la hipófisis y el consiguiente aumento de la producción de los precursores mencionados. Este incremento puede ser determinado analíticamente (17-hidroxicorticosteroides, 17-HOCS y esteroides -17cetogénicos, 17-KGS), como indicativo de la reactividad hipofisaria para la ACTH. La metirapona carece de actividad sobre la síntesis adrenocortical o gonadal de testosterona.
Básicamente, hay dos formas en que se utiliza la metirapona en diagnóstico del síndrome de Cushing; en la primera, que se suele realizar por la noche, se administra una única dosis de metirapona a las 11 pm, tomando la muestra de sangre a las 8 am para medir el nivel sérico de cortisol, de ACTH y de 11-desoxicortisol. En la segunda forma, se administra metirapona 6 veces al día durante 24 horas (cada 4 h) y posteriormente se recoge toda la orina emitida por el pacientes a lo lardo de 24 horas, determinándose la concentración de 17-hidroxicorticosteroides (17-OHCS); igualmente, se pueden tomar las muestras de sangre para medir el cortisol sérico, de ACTH y el 11-desoxicortisol.
Se consideran valores normales un aumento del 11-desoxicortisol en suero a más de 70 µg/L (0,2 µmol/L) de ACTH a más de 200 ng/L (44 pmol/L). Asimismo, un valor normal para el examen de orina de 24 horas muestra que la eliminación inicial de 17-OHCS urinario es más del doble. La falta de respuesta sugiere una insuficiencia adrenocortical secundaria; por su parte, un aumento excesivo de 17-OHCS y 17-KGS supone la existencia de síndrome de Cushing, es decir, que no existe un tumor adrenocortical capaz de producir cortisol de forma autónoma.
ASPECTOS MOLECULARES
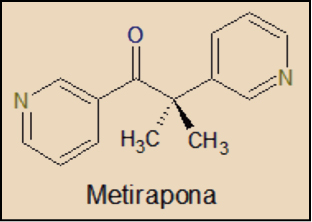
La metirapona es una molécula muy sencilla, que está estructuralmente relacionada con el ácido nicotínico (3-piridincarboxílico), cuyos derivados más conocidos son el NAD (dinucleótido de nicotinamida y adenina) y el NADP (nicotinamida adenina dinucleótido fosfato). Fue sintetizada por vez primera en 1958 y tan solo en 1961 ya había sido autorizada por la Food & Drug Administration (FDA), de Estados Unidos, para su uso en el diagnóstico del síndrome de Cushing.
EFICACIA Y SEGURIDAD CLÍNICAS
La sensibilidad y precisión diagnóstica, así como la eficacia clínica en el tratamiento del síndrome de Cushing, y la seguridad de la metirapona han sido determinadas fundamentalmente a partir de datos retrospectivos.
En relación al utilización al diagnóstico del síndrome de Cushing, en un estudio de cohorte retrospectivo (Avgerinos, 1994) que incluyó a 186 pacientes hospitalizados con diagnóstico clínico de síndrome de Cushing ACTH-dependiente, se les practicó las pruebas diagnósticas de la metirapona y de la dexametasona, para determinar sus respectivas sensibilidades, especificidades y precisiones diagnósticas. En el caso de la metirapona se determinaron la excreción urinaria de 17-hidroxicorticosteroides (17-OHCS) y los niveles plasmáticos de 11-desoxicortisol, mientras que en el caso de la prueba de la dexametasona, se determinaron los niveles urinarios de 17-OHCS y de cortisol libre. Del total de pacientes, 156 presentaban patología hipofisaria, 15 tenían secreción ectópica de ACTH y los restantes 15 no tuvieron diagnóstico tras la cirugía hipofisaria.
Tras la administración de metirapona, la combinación de los datos séricos y urinarios proporcionó un 71% (IC95% 62 a 79) de predicciones correctas, superior a la obtenida considerando exclusivamente los niveles de 17-OHCS (62%; IC95% 52 a 71) y los de 11-desoxicortisol (44%; IC95% 34 a 54); por su parte, la sensibilidad y especificidad de la prueba de la dexametasona fueron idénticas a la de la metirapona; sin embargo, combinando los criterios utilizados con metirapona con los de la prueba de la dexametasona, el porcentaje de predicciones correctas (82%; IC95% 75 a 87) fue superior que el obtenido por cada una de ellas por separado (p=0,001). Cuando ambos procedimientos analíticos se combinaron en la identificación del síndrome de Cushing hipofisario, la sensibilidad y la precisión diagnóstica aumentaron al 88% y 89%, respectivamente.
Por lo que se refiere a la utilización en el tratamiento del síndrome de Cushing, hay varias series retrospectivas de casos, pero no se dispone de ningún estudio prospectivo controlado con placebo con algún comparador activo (como el ketozonazol). El mayor y más reciente es un estudio multicéntrico que contiene datos retrospectivos procedentes de 13 hospitales universitarios de Gran Bretaña (Daniel, 2015b).
En total se recogieron en este estudio 195 pacientes con síndrome de Cushing, de las que 114 presentaban enfermedad de Cushing, 37 síndrome hipersecretor de ACTH ectópico, 43 enfermedad ACTK-independiente (10 con carcinoma adrenocortical, 30 con adenoma adrenal y 3 con hiperplasia adrenal ACTH-independiente), determinándose la curva diaria de niveles séricos de cortisol, los niveles séricos de cortisol a la 9 am y la cantidad total de cortisol libre en orina de 24 h; de los 195 pacientes, 164 (84%) recibieron solo metirapona. La media de edad de los pacientes era de 50 años y la mediana de duración del tratamiento fue de 3 meses.
Los resultados mostraron claras diferencias entre la primera y la última evaluación: 722,9 nmol/L (26,2 µg/dL) vs. 491,1 (17,8; p>0,0001) en la curva diaria de cortisol sérico; 882,9 nmol/L (32,0 µg/dL) vs. 348,6 (12,6; p>0,0001) en los niveles séricos de cortisol a las 9 am y 1483 nmol/24 h (537 µg/24 h) vs. 452,6 nmol/24 h (164 µg/24 h; p=0,003) en los niveles urinarios de cortisol libre en 24 h. Un 55% de los pacientes mantuvieron la curva diaria de cortisol sérico dentro de los límites considerados como objetivo (150-300 nmol/L), un 43% normalizaron la cantidad de cortisol libre en orina de 24 h, un 46% presentaban niveles séricos de cortisol a las 9 am ≤331 nmol/L (≤12,0 µg/dL) y un 76% ≤600 nmol/L (≤21,7 µg/dL).
La mediana de la dosis de metirapona utilizada en los pacientes con enfermedad de Cushing fue de 1375 mg, de 1500 mg en aquellos con síndrome ACTH ectópico, 750 mg para aquellos con enfermedad adrenal benigna y 1250 mg para los pacientes con carcinoma adrenocortical.
Desde el punto de vista de la seguridad, la metirapona en uso continuado presenta un perfil toxicológico relevante, habida cuenta de que tiende a incrementar los niveles de los precursores de cortisol y de aldosterona, afectando asimismo a la producción adrenocortical de testosterona, teóricamente favorecida al desviar el metabolismo esteroídico hacia la vía de la síntesis de andrógenos. En el estudio retrospectivo realizado en pacientes con síndrome de Cushing (Daniel, 2015b), se registraron eventos adversos en el 25% de los pacientes tratados, fundamentalmente de carácter gastrointestinal (náusea y vómitos) y mareos, usualmente dentro de las dos primeras semanas de iniciación del tratamiento o de incremento de la dosis, aunque todos ellos reversibles; otros eventos adversos frecuentes son sedación, cefalea y, más raramente, hipotensión, dolor abdominal, hirsutismo y dermatitis.
ASPECTOS INNOVADORES
La metirapona es un inhibidor de la síntesis de aldosterona y cortisol en la corteza suprarrenal, bloqueando de forma reversible y selectiva la 11β-hidroxilasa y, adicionalmente, la 18-hidroxilasa. El resultado es una reducción de los niveles de cortisol y, especialmente, de aldosterona. El medicamento ha sido autorizado como prueba diagnóstica para la insuficiencia de ACTH y en el diagnóstico diferencial del síndrome de Cushing ACTH-dependiente, así como para el manejo de pacientes con síndrome de Cushing endógeno.
En general, se considera que la prueba de la metirapona en el diagnóstico del síndrome de Cushing tiene una sensibilidad y especificidad muy similar a la prueba de supresión con altas dosis de dexametasona, con la ventaja de que la combinación de los resultados de ambas pruebas es aún más fiable que cada una de ellas por separado (Avgerinos, 1994), lo que viene a indicar un cierto grado de innovación incremental.
Por lo que respecta a su uso terapéutico en el síndrome de Cushing, los datos – aunque no proceden de estudios sistemáticos debidamente controlados – son lo suficientemente claros como para garantizar su utilidad, al menos en una proporción relevante de pacientes. En este sentido, un 55% de los pacientes mantuvieron la curva diaria de cortisol sérico dentro de los límites considerados como objetivo (150-300 nmol/L), un 43% normalizaron la cantidad de cortisol libre en orina de 24 h, un 46% presentaban niveles séricos de cortisol a las 9 am ≤331 nmol/L (≤12,0 µg/dL) y un 76% ≤600 nmol/L (≤21,7 µg/dL) (Daniel, 2015b).
Actualmente, la única alternativa farmacológica a la metirapona en el tratamiento del síndrome de Cushing es el ketoconazol, cuyo espectro de bloqueo enzimático es mucho más inespecífico que el de la metirapona, ya que actúa sobre diversos enzimas implicados en la síntesis de aldosterona, cortisol y testosterona a partir de colesterol (17α-hidrolasa, 20,22-desmolasa, 11β-hidroxilasa, 17,20-desmolasa y 18-hidroxilasa). Según los datos del mayor estudio retrospectivo realizado con ketoconazol en síndrome de Cushing (Castinetti, 2014), un 75% de los cuales respondió al tratamiento con dosis de 200 a 1.200 mg/día, normalizando en el 49% los niveles urinarios en 24 h de cortisol libre.
Aunque es evidente que la comparación de ambos estudios no puede tener ningún valor cuantitativo, sí al menos sugiere que metirapona y ketoconazol presentan una misma magnitud de efectos terapéuticos en el tratamiento del síndrome de Cushing. Por ello, la decisión de usar uno u otro, debe ser adoptada de forma personalizada, teniendo en cuenta sobre todo los potenciales efectos adversos del tratamiento. En este sentido, el hipogonadismo y la ginecomastia es un efecto bien documentado para el ketoconazol (debido al bloqueo de la síntesis adrenocortical de testosterona); por ello, parece razonable preferir la metirapona en pacientes varones jóvenes. Por su parte, la aparición de efectos androgénicos (hirsutismo) en las mujeres ha dado lugar a la suspensión del tratamiento en algunos casos y, por ello, el ketoconazol podría ser preferible en estas pacientes. Ambas opciones, más allá de estas particularidades, son aceptables en pacientes donde otras opciones, como la cirugía o la radioterapia, no sean aconsejables o no hayan producido los resultados buscados (Daniel, 2015a).
|
VALORACIÓN |
|
METIRAPONA  |
|
Grupo Terapéutico (ATC): V04CD. VARIOS. Otros agentes diagnósticos: pruebas de la función hipofisaria. |
|
Indicaciones autorizadas: Como prueba diagnóstica para la insuficiencia de ACTH y en el diagnóstico diferencial del síndrome de Cushing ACTH-dependiente. Para el manejo de pacientes con síndrome de Cushing endógeno. |
|
INNOVACIÓN moderada. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |
|
Novedad clínica: Posibilidad de asociar con otros tratamientos actualmente en vigor y útil en cuadros refractarios a los tratamientos actuales o en pacientes en los que el tratamiento estándar está contraindicado. |
|
Novedad molecular: Mecanismo de acción relativamente innovador, con un efecto más selectivo sobre la diana farmacológica. |
BIBLIOGRAFÍA
Bibliografía
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Ketoconazol de administración sistémica (comprimidos): suspensión de comercialización. Nota informativa MUH (FV), 21/2013, de 29 de julio de 2013. http://www.aemps.gob.es/informa/notasInformativas/medicamentosUsoHumano/seguridad/2013/NI-MUH_FV_21-2013-ketoconazol.htm
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Metopirone®. Sumario de características (ficha técnica). http://www.agemed.es/cima/dochtml/ft/79004/FichaTecnica_79004.html
- Avgerinos PC, Yanovski JA, Oldfield EH, Nieman LK, Cutler GB Jr. The metyrapone and dexamethasone suppression tests for the differential diagnosis of the adrenocorticotropin-dependent Cushing syndrome: a comparison. Ann Intern Med. 1994; 121(5): 318-27.
- Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008; 93(7): 2454-62. doi: 10.1210/jc.2007-2734.
- Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab. 2014; 99: 1623-30. doi:10.1210/jc.2013-3628
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Diagnóstico funcional farmacológico. En: Terapéutica farmacológica de los trastornos dermatológicos, oftalmológicos y otológicos. Agentes farmacológicos de diagnóstico. Madrid: Consejo General de Colegios Oficiales de Farmacéuticos; 2014. p. 267-98.
- Daniel E, Aylwin S, Mustafa O, Ball S, Munir A, Boelaert K, et al. Effectiveness of Metyrapone in Treating Cushing’s Syndrome: A Retrospective Multicenter Study in 195 Patients. J Clin Endocrinol Metab. 2015; 100(11): 4146-54. doi: 10.1210/jc.2015-2616.
- Daniel E, Newell-Price JD. Therapy of endocrine disease: steroidogenesis enzyme inhibitors in Cushing’s syndrome. Eur J Endocrinol. 2015; 172(6): R263-80. doi: 10.1530/EJE-14-1014.
- Espinosa de los Monteros Sánchez AL. Evaluación del paciente con problemas endocrinológicos. Pruebas bioquímicas en neuroendocrinología. Rev Endocrinol Nutr. 2005; 13(Supl.1): S7-S12
- Santos S, Santos E, Gaztambide S, Salvador J. Diagnóstico y diagnóstico diferencial del síndrome de Cushing. Endocrinol Nutr. 2009; 56(2): 71-84.
- Tresguerres Hernández JAF. Fisiopatología del eje hipotálamo-hipofisario. Fisiopatología de las suprarrenales. En: Principios de Fisiopatología para la Atención Farmacéutica. Módulo II. Consejo General de Colegios Oficiales de Farmacéuticos; Madrid, 2008; pp. 133-62.
- Verhelst JA, Trainer PJ, Howlett TA, Perry L, Rees LH, Grossman AB, et al. Short and long-term responses to metyrapone in the medical management of 91 patients with Cushing’s syndrome. Clin Endocrinol. 1991; 35: 169–78. doi: 10.1111/j.1365-2265.1991.tb03517.x
1 Además, dos hormonas secretadas por la placenta durante el embarazo, el lactógeno placentario y la gonadotropina coriónica, forman parte de las hormonas somatotrópicas y glucoproteicas, respectivamente.
Artículos relacionados
-
2 Abr 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
27 Feb 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
30 Dic 2024Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
