Trifluridina/Tipiracilo ▼(Servier) en cáncer colorrectal
 Nº400
Nº400

Resumen
La trifluridina es un agente antineoplásico de estructura nucleosídica que actúa como un antimetabolito de la base desoxitimidina, incorporándose a la cadena de ADN celular y provocando la formación de cadenas anómalas de ADN, las cuales determinan un efecto deletéreo la multiplicación celular. El medicamento está formado por la combinación de la trifluridina con tipiracilo (en proporción molar 1:0,5), un inhibidor del enzima timidina fosforilasa que responsable de la rápida inactivación de la trifluridina. El medicamento ha sido autorizado para el tratamiento de pacientes adultos con cáncer colorrectal metastásico que hayan sido previamente tratados o no se les considere candidatos al tratamiento con terapias disponibles, incluidas quimioterapia basada en fluropirimidinas, oxaliplatino e irinotecán, agentes anti-VEGF y agentes anti-EGFR. Los datos clínicos proceden de un amplio estudio realizado sobre 800 pacientes, encontrándose una mejora en la mediana de la supervivencia global de 2 meses con relación al placebo y de 0,3 meses en la supervivencia libre de progresión tumoral. Presenta un perfil toxicológico notable, en línea con otros antineoplásicos del grupo, destacando por encima de todos los efectos adversos de naturaleza hematológica y gastrointestinal, aunque el porcentaje de pacientes que suspendieron el tratamiento debido a los eventos adversos fue mayor con placebo que con el medicamento (10,6 vs. 13,6%). Visto en este contexto y con la prudencia que exige cualquier comparación indirecta de datos clínicos, su modesta eficacia es similar a la del regorafenib, aunque aparentemente con un grado algo mejor de tolerabilidad.
ASPECTOS FISIOPATOLÓGICOS
El cáncer colorrectal es el tipo de cáncer más comúnmente diagnosticado en España, representando el 15,0% de todos los nuevos casos, por delante del cáncer de próstata, el de mama y el de pulmón; esto supone en torno a 55 nuevos casos anuales por cada 100.000 varones españoles y unos 30 para las mujeres. Aproximadamente 15.500 personas fallecen en nuestro país por esta causa; solo el cáncer de pulmón, con 22.000 fallecimientos anuales, supera al cáncer colorrectal como causa de muerte de origen neoplásico. Afortunadamente, el cáncer colorrectal es el tumor que mejor se puede prevenir mediante un cribado poblacional relativamente sencillo y asequible, en el que la oficina de farmacia ya está jugando un papel de gran relevancia.
El tipo histológico más común de cáncer de colon es el adenocarcinoma (>93%), que puede ser mucinoso (coloide) o de células en anillo de sello; otros tipos histológicos mucho menos comunes son los escirrosos y los neuroendocrinos. Estos últimos suelen presentar un pronóstico más precario que las variantes de adenocarcinoma puro. Los adenocarcinomas constituyen también la gran mayoría de tumores rectales (>95%).
La localización más habitual de los tumores de colon son el descendente y sigma (40%), y el cecoascendente (40%); el restante 20% corresponde a colon transverso. Las tres cuartas partes de los tumores de recto se localizan en la ampolla, mientras que la cuarta parte restante se localiza en el recto sigmoide; solo un pequeño porcentaje (1-2%) se sitúan en el ano.
Las características fisiopatológicas y las sintomatologías del cáncer de colon y el de recto son muy similares, motivo por el cual se consideran habitualmente como una misma entidad patológica, con escasas diferencias. Los síntomas son, en general, poco específicos y consisten fundamentalmente en hemorragia rectal (sangre fresca en las heces o pérdidas a través del ano), cambios de hábitos intestinales, dolor abdominal, obstrucción intestinal, cambios en el apetito, pérdida de peso y debilidad general.
Con excepción de los síntomas obstructivos, los síntomas no se correlacionan necesariamente con el estadio de la enfermedad ni con la localización del tumor. Sin embargo, la localización tiene importantes consecuencias prácticas, ya que, en el caso del cáncer rectal, la distancia del tumor desde la musculatura del esfínter anal es determinante sobre la capacidad de llevar a cabo cirugías para preservar el esfínter anal. Asimismo, las restricciones óseas de la pelvis limitan el acceso quirúrgico al recto y ello resulta en una probabilidad menor de obtener márgenes ampliamente negativos y un riesgo más alto de recidiva local.
Aproximadamente el 75% de todos los casos de cáncer colorrectal son esporádicos y los restantes se relacionan con la historia familiar y/o enfermedades intestinales inflamatorias; de los casos familiares, solo el 5% tienen un patrón de herencia bien definido.
Existen dos mecanismos génicos independientes que pueden conducir al desarrollo del cáncer colorrectal. El primero es iniciado por la inactivación mutacional del gen supresor de tumor APC, el cual es responsable de la poliposis adenomatosa familiar (FAP) y de aproximadamente el 85% de los cánceres colorrectales esporádicos. Algunos de estos carcinomas se desarrollan tras la activación mutacional de β-catenina (CTNNB1), cuya actividad normalmente regulada por APC. El segundo mecanismo es iniciado a través de la inactivación de una familia de genes supresores de tumor involucrados en la reparación del daño al ADN, conocidos como genes MMR o Mismatch (MSH2, MLH1 y PMS2). Se han encontrado mutaciones en estos genes tanto en individuos con cáncer colorrectal esporádico (15%) como hereditario. Se ha descrito además inactivación mutacional de otros genes supresores de tumor como DPC4/SMAD4 y activación mutacional de oncogenes como COX-2, los cuales están presentes en etapas tardías de la formación del cáncer colorrectal.
En resumen, el cáncer colorrectal evidencia una gran heterogeneidad genética, debido a que puede desarrollarse por diferentes vías, dependiendo del gen alterado inicialmente; por ejemplo, si ocurre una alteración en un gen supresor de tumores o en un protooncogén, como APC o K-RAS respectivamente, se desarrolla la vía supresora; si, por el contrario, la mutación se presenta en un gen de reparación como MLH1 o MSH2, se desencadena la vía mutadora, mientras que si se produce una inactivación en la expresión de genes por mecanismos epigenéticos, el cáncer se podría desarrollar por la vía de la metilación.
El cáncer de colon es una enfermedad altamente tratable y, a menudo, curable cuando se localiza en el intestino. La cirugía es la forma primaria de tratamiento y resulta en la curación de aproximadamente 50% de los pacientes, aunque la recidiva es un problema grave y, con frecuencia, la causa definitiva de muerte. Sea como fuere, la cirugía es la primera y única opción en los estadios O (cáncer in situ) a II, también se emplea en la fase III pero acompañada de quimioterapia adyuvante; en la fase IV, las metástasis hepáticas son tratadas con cirugía y quimioterapia neoadyuvante, mientras que los cuadros de cáncer recidivante son tratados, una vez más, con cirugía, quimioterapia y terapia dirigida.
El tratamiento del cáncer de recto varía en algunos aspectos del de cáncer de colon debido al aumento del riesgo de recidiva local y un pronóstico general más precario. Las diferencias incluyen la técnica quirúrgica, el uso de radioterapia y el método de administración de la quimioterapia. Además de determinar cuál es la intención de la cirugía del cáncer de recto (es decir, curativa o paliativa), es importante considerar aspectos terapéuticos relacionados con el mantenimiento o el restablecimiento de la normalidad del esfínter anal, el funcionamiento genitourinario y la función sexual.
El valor potencial de la quimioterapia adyuvante para los pacientes de cáncer de colon en estadio II es polémico. Los análisis conjuntos y los metanálisis indican una mejora leve de la supervivencia global de los pacientes tratados con terapia basada en fluorouracilo (5-FU) adyuvante. Antes del año 2000, este fármaco era la única quimioterapia citotóxica útil en el entorno adyuvante para los pacientes de cáncer de colon en estadio III; sin embargo, ya en el año 2000, se estableció que la capecitabina era preferible en términos de eficacia y de seguridad, siendo al menos equivalente a la combinación de fluorouracilo y leucovorina (ácido folínico). Asimismo, la combinación del oxaliplatino al fluororacilo y leucovorina mejoró la supervivencia global de esta combinación; el tegafur (UFT), un análogo del fluorouracilo, también ha sido utilizado. Por su parte, el irinotecán, utilizado en el cáncer de colon metastásico, no es útil en tumores localizados. Inicialmente se utilizaron combinaciones de fluorocilo con ácido folínico (leucovorina, LV) en diversos esquemas (bolo o infusión continua), con un mejor perfil de toxicidad en aquellos de infusión continua. Las fluoropirimidinas orales (capecitabina, UFT) han demostrado ser al menos tan eficaces como las combinaciones de 5-FU y LV, con la ventaja de una administración más cómoda. Posteriormente, se demostró que la adición de oxaliplatino a los esquemas de 5-FU y LV (esquema FOLFOX) aumentaba la supervivencia libre de enfermedad y la supervivencia global frente a esquemas con 5-FU y LV. La combinación de capecitabina y oxaliplatino (esquema CAPOX) también se ha mostrado superior a 5-FU y LV. Por ello, el estándar de tratamiento adyuvante actualmente consiste en 6 meses de quimioterapia mediante la pauta FOLFOX, o bien CAPOX como alternativa.
Hasta un cuarto de los pacientes con tumores de colon presentan metástasis (las más frecuentes, hepáticas y pulmonares) en el momento del diagnóstico, a pesar de lo cual, puede ser subsidiario de cirugía: si el tumor primario causa problemas (obstrucción, sangrado, etc.) puede realizarse, en el caso de que sea posible, una extirpación del mismo, o una cirugía paliativa sin resección tumoral para restituir el tránsito intestinal (colostomía de descarga). Por el momento, no existe tratamiento curativo para el cáncer colorrectal metastásico, aunque puede prolongarse la vida del paciente de forma significativa, pasando de 6 meses (supervivencia media sin tratamiento) hasta un promedio de 20 con quimioterapia selectiva. Para los pacientes de enfermedad recidivante local o metastásica del hígado solamente o del pulmón solamente, la resección quirúrgica, si es posible, es el único tratamiento potencialmente curativo.
En la actualidad, hay un grupo de fármacos autorizados para el tratamiento de pacientes de cáncer colorrectal metastásico que se usan solos o en combinación. Además de los fármacos ya comentados previamente (fluoropirimidinas, irinotecán y oxaliplatino), son útiles en el tratamiento de la enfermedad metastásica determinados anticuerpos monoclonales. En este sentido, se ha observado que la supervivencia de los pacientes es mayor cuantos más fármacos lleguen a recibir a lo largo de la evolución de la enfermedad. Por ello, lo habitual es administrar combinaciones de quimioterapia (fluoropirimidina combinada con oxaliplatino o irinotecán) y cuando la enfermedad progresa a un tratamiento concreto, se cambia de esquema de quimioterapia. Se han diseñado múltiples esquemas de tratamiento. Algunos de ellos (FOLFOX y CAPOX) ya se han mencionado para el tratamiento de la enfermedad localizada, pero otros, basados en el uso de irinotecán combinado con 5-FU (FOLFIRI, IFL), sólo son útiles en la enfermedad metastásica.
Para los pacientes con cáncer de recto metastásico que recidiva localmente, solo en el hígado o solo en el pulmón, la resección quirúrgica es el único tratamiento potencialmente curativo. Los pacientes con metástasis pulmonar limitada y los pacientes con metástasis tanto pulmonares como hepáticas, también se pueden considerar para resección quirúrgica, con una supervivencia a 5 años posible en pacientes muy seleccionados. El uso de la quimiorradioterapia de inducción para pacientes no irradiados previamente con recidivas pelvianas localizadas en estado avanzado (compromiso de la pared pelviana lateral, el sacro o los órganos adyacentes) puede aumentar la resecabilidad y permitir la preservación del esfínter.
El desarrollo de nuevos anticuerpos monoclonales selectivos está incrementando las opciones terapéuticas disponibles y, en particular, el cáncer colorrectal metastásico es uno de los tumores donde el uso de estos fármacos está más desarrollado. En general, se utilizan combinándolos con la quimioterapia convencional antes mencionada.
El bevacizumab es un anticuerpo monoclonal parcialmente humanizado que se une al factor de crecimiento endotelial vascular. Actúa, por tanto, sobre la neovascularización tumoral. En combinación con quimioterapia basada en irinotecán (FOLFIRI o IFL), ha demostrado mejorar los resultados de la quimioterapia sola, aumentando la supervivencia global de los pacientes. También existen datos favorables en la combinación con esquemas con oxaliplatino (CAPOX o FOLFOX), retrasando la progresión de la enfermedad.
El cetuximab es un anticuerpo monoclonal parcialmente humanizado contra el receptor del factor de crecimiento epidérmico (EGFR) y, por tanto, tiene como objetivo fundamental frenar la angiogénesis tumoral. Debido a que el cetuximab afecta la señalización de la tirosina cinasa en la superficie de la membrana celular, los tumores con mutaciones que causan activación de las vías descendientes del EGFR de la mutación K-RAS, no son sensibles a sus efectos ya que de existir mutaciones de K-RAS, ésta se encuentra activada de manera constante e independiente del EGFR, por lo que su bloqueo por cetuximab no resulta útil.
Otro fármaco anti-EGFR usado en estos pacientes es el panitumumab, que requiere para su uso, como en el caso anterior, descartar la existencia de mutación de K-RAS. Está admitido su uso en monoterapia tras progresión a primera línea. Los efectos secundarios son muy similares a los del cetuximab, excepto por una muy baja incidencia de reacciones de hipersensibilidad, por lo que no precisa premedicación.
El aflibercept es una proteína de fusión recombinante, formada por porciones de los lugares de unión del factor de crecimiento del endotelio vascular (VEGF) de los dominios extracelulares de los receptores humanos VEGFR-1 y 2, fusionados con una porción Fc de la IgG1 humana. El fármaco actúa como un receptor soluble que se une al VEGF-A, actuando de señuelo, con una afinidad más alta que sus receptores nativos, con lo que previene la unión de los ligandos endógenos a dichos receptores y, de esta forma, bloquea la señalización correspondiente. Para el cáncer colorrectal se utiliza en combinación con quimioterapia con irinotecán-fluorouracilo-ácido folínico (FOLFIRI).
Aunque no es un medicamento biológico, el regorafenib se incluye en este campo por estar estrechamente relacionado con los mecanismos farmacológicos de la inmunoquimioterapia del cáncer colorrectal. Se trata del primer inhibidor de tirosina cinasas (TKI) comercializado que ha conseguido demostrar algún grado de beneficio clínico, en términos de supervivencia en los pacientes con cáncer colorrectal metastásico con progresión tumoral pese a haber sido intensamente tratados con los medicamentos de referencia actuales. El regoranib es un inhibidor de un amplio grupo de tirosina cinasas, incluyendo las que forman parte de receptores de ligandos endógenos implicados en la angiogénesis tumoral (VEGFR1, 2 y 3; TIE2, FGFR1 Y PDGFRB), la oncogénesis (KIT, RET, RAF-1, BRAF) y el microambiente tumoral (PDGFR, FGFR). El medicamento ha sido autorizado para el tratamiento de pacientes adultos con cáncer colorrectal metastásico que han sido previamente tratados con las terapias disponibles o no se les considera candidatos adecuados a dichas terapias, incluyendo quimioterapia basada en fluoropirimidinas, terapia anti-VEGF y terapia anti-EFGR; también está indicado en pacientes adultos con tumores del estroma gastrointestinal (GIST) irresecables o metastásicos que progresaron durante el tratamiento previo con imatinib y sunitinib o son intolerantes al mismo (Cuéllar, 2016).
ACCIÓN Y MECANISMO
La trifluridina es un agente antineoplásico de estructura nucleosídica que actúa como un antimetabolito de la base desoxitimidina, incorporándose a la cadena de ADN celular y provocando la formación de cadenas anómalas de ADN, las cuales determinan un efecto deletéreo la multiplicación celular. El medicamento está formado por la combinación de la trifluridina con tipiracilo (en proporción molar 1:0,5), un inhibidor del enzima timidina fosforilasa que responsable de la rápida inactivación de la trifluridina; en consecuencia, el tipiracilo permite mantener niveles adecuados de trifluridina para que ésta ejerza su efecto citostático; de hecho, la coadministración de tipiracilo incrementa en 37 veces la exposición celular a la trifluridina. El medicamento ha sido autorizado para el tratamiento de pacientes adultos con cáncer colorrectal metastásico que hayan sido previamente tratados o no se les considere candidatos al tratamiento con terapias disponibles, incluidas quimioterapia basada en fluropirimidinas, oxaliplatino e irinotecán, agentes anti-VEGF y agentes anti-EGFR.
Aunque está estrechamente relacionada con el fluorouracilo, la trifluridina ejerce su efecto citotóxico a través de un mecanismo algo diferente, ya que el fluorouracilo es transformado en flurouridina-monofosfato, que es fosforilado posteriormente a difosfato. Tras ello, se forma el correspondiente 2-desoxinucleótido por acción del enzima ribonucleótido reductasa, siendo posteriormente reducido a la forma de monofosfato (5-fluoro-2-desoxiuridina-monofosfato, 5-FdUMP). Éste es precisamente el auténtico antimetabolito o forma activa del fluorouracilo, ya que es capaz de inhibir la síntesis de timidilato y, con ella, la de ADN, como consecuencia de la formación de un complejo ternario estable entre 5-FdUMP, la timidilato sintasa y el cofactor N5,10-metileno-tetrahidrofolato.
La trifluridina presenta actividad antiviral, actuando también como antimetabolito de la timidina en la síntesis de ADN viral. Actúa sobre virus del Herpes simplex, tipos 1 y 2.
ASPECTOS MOLECULARES
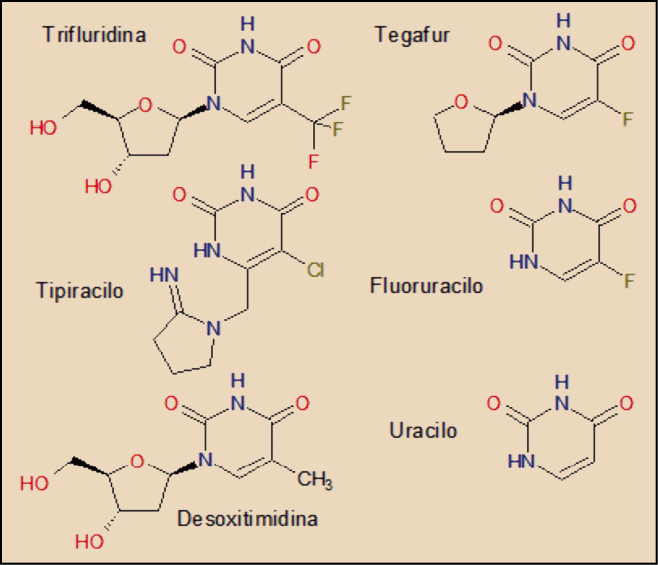
La trifluridina es un evidente análogo molecular de la desoxitimidina, una de las bases nucleicas del ADN, de la que tan solo se diferencia en que el grupo metilo (-CH3) del anillo pirimidínico está sustituido por otro de trifluorometilo (-CF3) en la trifluridina.
La analogía molecular de ambos con el tiperacilo es también evidente, lo cual se justifica obviamente por el hecho de que es un potente inhibidor del enzima timidina fosforilasa, capaz de desactivar químicamente a la trifluridina de forma rápida.
Aunque el fluorouracilo guarda una estrecha relación estructural con la trifluridina, su mecanismo de acción citotóxico es algo diferente, como ya se ha indicado.
EFICACIA Y SEGURIDAD CLÍNICAS
La eficacia y la seguridad clínicas de la combinación trifluridina/tipiracilo han sido adecuadamente contrastadas en las indicaciones autorizadas mediante un ensayo clínico de fase 3 (confirmatorio de eficacia y seguridad), aleatorizado, multicéntrico, doblemente ciego y controlado con placebo (RECOURSE; Mayer, 2015).
El estudio fue realizado sobre un total de 800 pacientes con adenocarcinoma de colon o de recto histológica o citológicamente confirmado, que hubieran recibido al menos dos regímenes quimioterápicos para el cáncer colorrectal metastásico y que fueran refractarios o que no hubiesen tenido una respuesta satisfactoria a regímenes conteniendo fluoropirimidinas, irinotecán, oxaliplatino, algún anticuerpo monoclonal anti-VEGF (como bevacizumab) y, para aquellos pacientes con genotipo KRAS salvaje (no mutado), algún anticuerpo monoclonal anti-EGFR (cetuximab, panitumumab).
Todos los pacientes recibieron el mejor tratamiento de soporte y aleatoriamente fueron asignados a recibir por vía oral dos dosis diarias de placebo o de trilfuridina/tipiracilo (35 mg/m2/dosis, en proporción 1:0,5) durante cinco días a la semana, con dos días de descanso, durante las dos primeras semanas, seguidas de otras dos semanas de descanso, completando así ciclos mensuales (cuatro semanas), que se mantuvieron hasta la progresión de la enfermedad o toxicidad intolerable por el paciente.
En cuanto a las características demográficas y clínicas de los pacientes, un 61% eran varones, la mediana de edad era de 64 años y la proporción de pacientes con genotipo KRAS no mutado (salvaje, wild) era de 49%; la localización del tumor primario era en el colon en un 62% de los pacientes, y en el recto en el restante 38%, con una mediana de tiempo transcurrido desde la confirmación de metástasis hasta el inicio del estudio de 31 meses.
Como variable primaria de eficacia se utilizó la supervivencia global, definida como el tiempo transcurrido desde el inicio del estudio hasta la fecha de la muerte. Como variables secundarias se usaron la supervivencia libre de progresión tumoral, definida como el tiempo transcurrido entre el inicio del estudio hasta la progresión tumoral determinada radiológicamente o la muerte por cualquier causa; el tiempo hasta el fallo del tratamiento (desde el inicio del estudio hasta progresión tumoral, descontinuación del tratamiento o muerte por cualquier causa), tasa de respuesta global (proporción de pacientes con evidencia de respuesta completa o parcial) y tasa de control de la enfermedad (tasa de pacientes con respuesta completa o parcial, o estabilización de la enfermedad).
Los resultados obtenidos mostraron como supervivencia global una mediana de 7,2 meses con trifluridina/tipiracilo vs. 5,2 meses con placebo (tasa de riesgo, HR=0,69; IC95% 0,59 a 0,81; p<0,0001), con una tasa de supervivencia a un año del 27,1 vs. 16,6% de los pacientes. Por lo que respecta a la supervivencia libre de progresión tumoral, la mediana fue de 2,0 vs. 1,7 meses (HR=0,48; IC95% 0,41 a 0,57; p<0,0001), la mediana del tiempo hasta el fallo del tratamiento fue de 1,9 vs. 1,7 meses (HR=0,50; IC95% 0,42 a 0,58; p<0,0001), la tasa de respuesta global del 1,6 vs. 0,4% (p<0,0001) y la tasa de control de la enfermedad del 44,0 vs. 16,3% (p<0,0001).
Cuando el análisis de los resultados se circunscribe a los pacientes españoles (n=112; media de 61 años, 62% varones) del estudio anterior, los resultados fueron homogéneos con los generales, con tasas de supervivencia global de 6,6 vs. 4,6 meses (HR=0,47; IC95% 0,28 a 0,78; p=0,0032) y de supervivencia libre de progresión tumoral de 2,0 vs. 1,7 meses (HR=0,47; IC95% 0,30 a 0,74; p=0,001) (Longo Muñoz, 2016).
Desde el punto de vista de la seguridad, la combinación trifluridina/tipiracilo presenta un perfil toxicológico notable, en línea con otros antineoplásicos del grupo, destacando por encima de todo los efectos adversos de naturaleza hematológica y gastrointestinal. La incidencia de global de eventos adversos relacionados con el tratamiento fue del 86% con trifluridina/tipiracilo vs. 55% con placebo, la de eventos adversos graves (grado ≥3) fue del 69 vs. 52% y el porcentaje de pacientes que suspendieron el tratamiento debido a los eventos adversos fue del 10,6 vs. 13,6% (mayor con placebo).
Los eventos adversos más comunes que aparecieron durante el tratamiento fueron anemia (32 vs. 4,5%), neutropenia (29 vs. 0%), diarrea (24 vs. 9,1%), náusea (39 vs. 11%), vómitos (20 vs. 4,5%), astenia (11 vs. 4,5%), fatiga (25 vs. 10%) y anorexia (27 vs. 11%). En cuanto a los eventos adversos más graves (grado ≥3), los más comunes fueron anemia (12,2 vs. 1,9%), neutropenia (20,1 vs. 0%), diarrea (2,3 vs. 0%) y fatiga (2,1 vs. 1,9%).
ASPECTOS INNOVADORES
La trifluridina es un agente antineoplásico de estructura nucleosídica que actúa como un antimetabolito de la base desoxitimidina, incorporándose a la cadena de ADN celular y provocando la formación de cadenas anómalas de ADN, las cuales determinan un efecto deletéreo la multiplicación celular. El medicamento está formado por la combinación de la trifluridina con tipiracilo (en proporción molar 1:0,5), un inhibidor del enzima timidina fosforilasa que responsable de la rápida inactivación de la trifluridina. El medicamento ha sido autorizado para el tratamiento de pacientes adultos con cáncer colorrectal metastásico que hayan sido previamente tratados o no se les considere candidatos al tratamiento con terapias disponibles, incluidas quimioterapia basada en fluropirimidinas, oxaliplatino e irinotecán, agentes anti-VEGF y agentes anti-EGFR.
Los datos clínicos proceden de un amplio estudio realizado sobre 800 pacientes, encontrándose una mejora en la mediana de la supervivencia global de 2 meses (7.2 vs. 5,2) con relación al placebo y de 0,3 meses (2,0 vs. 1,7) en la supervivencia libre de progresión tumoral, con una tasa de supervivencia a un año del 27,1 vs. 16,6%. Entre los 112 pacientes españoles incluidos en el estudio, los resultados fueron homogéneos con los generales.
La combinación trifluridina/tipiracilo presenta un perfil toxicológico notable, en línea con otros antineoplásicos del grupo, destacando por encima de todo los efectos adversos de naturaleza hematológica y gastrointestinal, aunque el porcentaje de pacientes que suspendieron el tratamiento debido a los eventos adversos fue mayor con placebo que con el medicamento (10,6 vs. 13,6%).
No se dispone de estudios directamente comparativos con otros fármacos indicados en cáncer colorrectal metastásico en pacientes que previa e infructuosamente hayan sido sometidos a varios regímenes quimio e inmunoterápicos. De hecho, estos pacientes tienen muy pocas opciones entre el arsenal farmacológico actualmente disponible, destacando como única excepción el regorafenib.
El regoranib es un inhibidor de un amplio grupo de tirosina cinasas (TK), incluyendo las que forman parte de receptores de ligandos endógenos implicados en la angiogénesis tumoral (VEGFR1, 2 y 3; TIE2, FGFR1 Y PDGFRB), la oncogénesis (KIT, RET, RAF-1, BRAF) y el microambiente tumoral (PDGFR, FGFR). En pacientes con cáncer colorrectal metastásico progresivo tras los tratamientos estándar actuales, ha mostrado incrementos en torno a 1,4 meses de la supervivencia global, con tasas de supervivencia a los 12 meses del 24 vs. 17% frente al placebo. Presenta una elevada incidencia de efectos adversos (debilidad, cansancio, anorexia, síndrome mano-pie, diarrea, infecciones, hipertensión y disfonía, etc.), que obliga a tratamientos adicionales, a frecuentes ajustes posológicos (más de la mitad de los pacientes) o incluso a la suspensión definitiva del tratamiento (2-8%) (Cuéllar, 2015).
Visto en este contexto y con la prudencia que exige cualquier comparación indirecta de datos clínicos, la modesta eficacia de la combinación trifluridina/tipiracilo (algo explicable, dada la condición terminal de este tipo de pacientes) puede equipararse a la del regorafenib, aunque con un grado de tolerabilidad algo mejor para trifluridina/tipiracilo.
|
VALORACIÓN |
|
TRILFURIDINA/TIPIRACILO
|
|
Grupo Terapéutico (ATC): L01BC. AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES. Antimetabolitos: análogos de pirimidina. |
|
Indicaciones autorizadas: Tratamiento de pacientes adultos con cáncer colorrectal metastásico que hayan sido previamente tratados o no se les considere candidatos al tratamiento con terapias disponibles, incluidas quimioterapia basada en fluropirimidinas, oxaliplatino e irinotecán, agentes anti-VEGF y agentes anti-EGFR. |
|
INNOVACIÓN MODERADA. Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar |

|
FÁRMACOS RELACIONADOS REGISTRADOS EN ESPAÑA |
|||
|
Fármaco |
Medicamento® |
Laboratorio |
Año |
|
Fluorouracilo |
Fluoro Uracil |
Meda Pharma |
1964 |
|
Tegafur |
Utefos |
Mylan (Almirall) |
1978 |
|
Trifluridina/Tipiracilo |
Lonsurf |
Servier |
2017 |
BIBLIOGRAFÍA
Bibliografía
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Informe de Posicionamiento Terapéutico de trifluridina/tipiracil hidrocloruro (Lonsurf®) en cáncer colorrectal. IPT, 58/2016. V1 (28 de noviembre de 2016). https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/IPT-trifluridina-Lonsurf-cancer-colorrectal.pdf
- Burness CB, Duggan ST. Trifluridine/Tipiracil: A Review in Metastatic Colorectal Cancer. Drugs. 2016; 76(14): 1393-402. doi: 10.1007/s40265-016-0633-9.
- Consejo General de Colegios Oficiales de Farmacéuticos. Bot PLUS WEB. https://botplusweb.portalfarma.com/
- Cuéllar Rodríguez S. Cáncer colorrectal. Panorama Actual Med 2016; 40(390): 5-24.
- Cuéllar Rodríguez S. Regorafenib (Stivarga®) en cáncer colorrectal y GIST. Panorama Actual Med 2015; 39(382): 289-94.
- European Medicines Agency (EMA). Lonsurf®. European Public Assessment Report (EPAR). EMA/165020/2016; EMEA/H/C/003897. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003897/WC500206248.pdf
- Longo-Muñoz F, Argiles G, Tabernero J, Cervantes A, Gravalos C, Pericay C, et al. Efficacy of trifluridine and tipiracil (TAS-102) versus placebo, with supportive care, in a randomized, controlled trial of patients with metastatic colorectal cancer from Spain: results of a subgroup analysis of the phase 3 RECOURSE trial. Clin Transl Oncol 2016.doi:10.1007/s12094-016-1528-7.
- Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, García-Carbonero R, Mizunuma N, et al; RECOURSE Study Group. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015; 372(20): 1909-19. doi: 10.1056/NEJMoa1414325.
Artículos relacionados
-
5 Dic 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
29 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
-
6 Oct 2025Medicamentos con nuevos principios activos o biosimilares Medicamentos con nuevos principios activos o biosimilares
