Selumetinib (▼Koselugo®) en neurofibromatosis tipo I
 Nº467
Nº467

Resumen
Selumetinib es un nuevo inhibidor oral, selectivo y no competitivo con ATP, de las proteínas cinasa 1 y 2 activadas por mitógenos (MEK1/2), componentes críticos de la ruta de señalización bioquímica RAF-MEK-ERK regulada por RAS, la cual se activa con frecuencia en los cánceres humanos por una variedad de tipos de receptores y estimula la proliferación de las células tumorales y su supervivencia. La actividad inhibitoria del fármaco detiene la señalización por la vía RAF-MEK-ERK, lo que se traduce en un efecto antiproliferativo de células tumorales como las de los neurofibromas (deficitarias de la actividad inhibitoria de neurofibromina-1 sobre RAS). El medicamento, designado como huérfano, ha recibido una autorización condicional de comercialización para el tratamiento de pacientes pediátricos de 3 años en adelante con neurofibromatosis de tipo 1 (NF1) que presenten neurofibromas plexiformes (NFP) sintomáticos e inoperables.
Su autorización se ha sustentado en los datos de un estudio de fase 1/2 de diseño abierto, multicéntrico y de un solo grupo (n= 50), aún en marcha y cuya variable principal de eficacia fue la tasa de respuesta objetiva (TRO), definida como la suma de respuestas completas –desaparición del NFP diana– y de respuestas parciales –reducción ≥ 20% del volumen del NFP tras 3-6 meses en comparación con el valor basal–. Los datos más actualizados reflejan una TRO del 68%, en todos los casos respuestas parciales; la mediana del cambio óptimo respecto al inicio fue de -28% del tamaño del NFP y ninguno de los pacientes tratados experimentó progresión de la enfermedad. Tras un seguimiento superior a 41 meses, no se alcanzó la mediana de duración de la respuesta, que en casi 2 de cada 3 pacientes respondedores (62%) perduró más allá de los 3 años.
Con respecto a la seguridad del tratamiento, los efectos tóxicos más frecuentes fueron náuseas, vómitos o diarrea, un aumento asintomático en el nivel de creatinfosfocinasa, erupción acneiforme y paroniquia. El perfil toxicológico del nuevo fármaco se considera aceptable y clínicamente manejable, pues la incidencia de eventos adversos de grado 3 es baja (3%), siendo los más frecuentes la diarrea, anemia, fiebre y aumento de creatinfosfocinasa en sangre.
Selumetinib incorpora un mecanismo de acción novedoso en su indicación, para la que no existe actualmente ninguna alternativa farmacológica aprobada.
El tratamiento erradicador de los NFP, que afectan aproximadamente a la mitad de pacientes con NF1, se basa en la cirugía, pero este abordaje no siempre es una opción debido a la posibilidad de complicaciones. Los datos disponibles de eficacia en niños y adolescentes apuntan a una mejora en aspectos como la calidad de vida, la intensidad de dolor o las capacidades motoras. Sin embargo, la eficacia del fármaco podrá ser variable en los distintos pacientes dada la heterogeneidad de las manifestaciones, y por ahora se desconoce si el fármaco incidirá sobre el curso natural de la enfermedad o si afectará al desarrollo de los niños. Existe asimismo incertidumbre sobre su seguridad a largo plazo y sobre la evolución de los tumores si se discontinúa el tratamiento. A pesar de que se requiere de datos más exhaustivos, selumetinib se erige como la única alternativa terapéutica a la cirugía en casos considerados inoperables, pudiendo facilitar su administración una resección quirúrgica posterior.
Aspectos fisiopatológicos
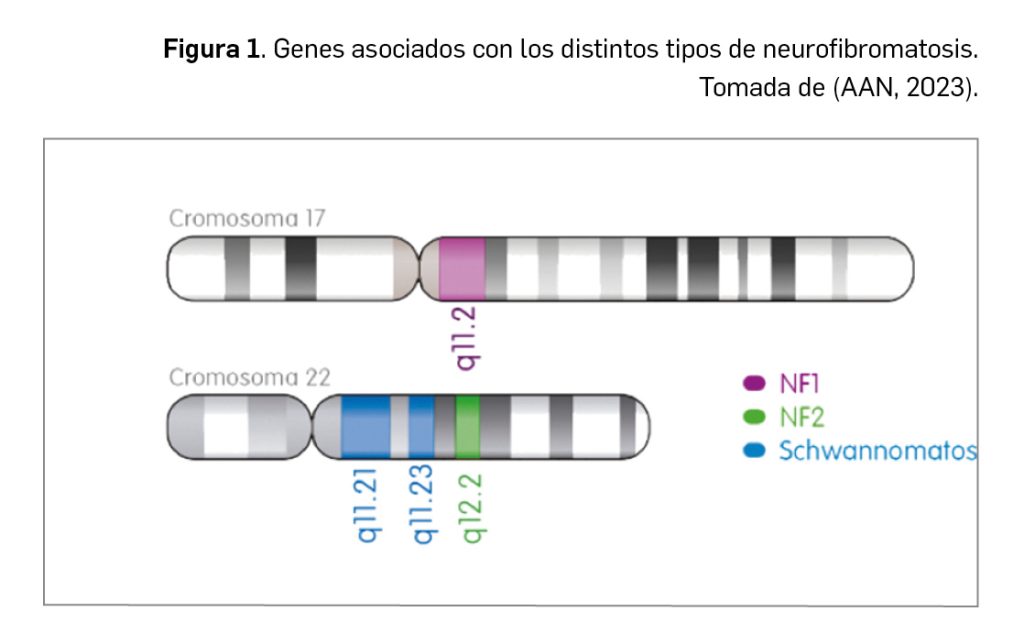
Las neurofibromatosis son un grupo de enfermedades genéticas raras caracterizadas por una predisposición al desarrollo de tumores benignos alrededor de los nervios y en la piel. A grandes rasgos, se distinguen tres tipos, cada una asociada con un defecto genético concreto (Figura 1) y con distintas características clínicas:
- Neurofibromatosis tipo 1: por lo general aparece en la infancia, con signos que suelen ser evidentes antes de los 10 años. Las manifestaciones cutáneas, neurológicas, esqueléticas y neoplásicas suelen ser progresivas y de intensidad leve a moderada, pudiendo llegar a graves.
- Neurofibromatosis tipo 2: mucho menos frecuente, los signos y síntomas aparecen como resultado del desarrollo de tumores benignos que crecen lentamente en los dos oídos.
- Schwannomatosis: es un tipo raro de neurofibromatosis que normalmente afecta a las personas adultas, después de los 20 años, y se caracteriza por la formación de tumores en los nervios craneales, espinales y periféricos.
En conjunto, son enfermedades hereditarias, con herencia autosómica dominante: si uno de los progenitores esté afectado, la probabilidad de que cada uno de los hijos padezca la patología es del 50%. Se trata, de hecho, de unas de las enfermedades autosómicas dominantes más comunes en los seres humanos. No obstante, hasta la mitad de los casos de neurofibromatosis diagnosticados son debidos a mutaciones espontáneas “de novo”.
De forma particular, la neurofibromatosis tipo I (en adelante, NF1) se debe a mutaciones germinales o microdeleciones del gen supresor de tumores NF1, situado en el cromosoma 17, que codifica para la proteína supresora de tumores neurofibromina 1. Esta proteína actúa en condiciones fisiológicas como activadora de la guanosina 5’ trifosfato (GTP)asa, la cual promueve a su vez la conversión de RAS-GTP activo en guanosina 5’-difosfato RAS inactivo, o sea, neurofibromina 1 actúa como un regulador negativo del protooncogén RAS, una molécula de señalización clave en el control del crecimiento celular.
La mutación de NF1 conducente a la pérdida de función de la proteína da como resultado un defecto en la inactivación de RAS, que estará constitutivamente activado. Los pacientes con NF1 comienzan su vida con 1 copia mutada (no funcional) y 1 copia funcional del gen en cada célula de su organismo, lo que determina que, aunque muchas de las características clínicas de la enfermedad son evidentes desde el nacimiento, se necesita una pérdida completa de la función del gen –mediante la aparición de una mutación somática en NF1 en células seleccionadas– para la aparición de tumores.
Ciertos estudios epidemiológicos han estimado cifras de prevalencia global de NF1 entre 20 y 24 casos por cada 100 000 habitantes, si bien los trabajos centrados en población pediátrica o adolescente apuntan a cifras ligeramente superiores, entre 18 y 34 casos por cada 100 000 habitantes en todo el mundo.
Desde el punto de vista clínico, los pacientes con NF1 tienen un mayor riesgo de desarrollar tumores del sistema nervioso central y periférico. Los neurofibromas plexiformes (en adelante, NFP) son uno de los tumores benignos más comunes y ocurren aproximadamente entre el 20% y el 50% de los pacientes que sufren la enfermedad. En cambio, los tumores malignos de la vaina del nervio periférico (TMVNP) a menudo surgen en NFP preexistentes y, siendo extremadamente raros en la población general, se estima que el riesgo de que se desarrollen a lo largo de la vida de un paciente con NF1 es del 8% al 16%, con una incidencia anual del 4,6% entre pacientes con NF1 (vs. 0,0001% en población general). Otros tumores asociados con la enfermedad incluyen: gliomas de bajo grado (los gliomas de la vía óptica ocurren en ~15% de los pacientes), así como tumores malignos como gliomas de alto grado, cáncer de mama, leucemia, feocromocitomas y tumores del estroma gastrointestinal.
En general, los neurofibromas son tumores de la vaina nerviosa histológicamente benignos, que pueden clasificarse ampliamente en: neurofibromas dérmicos y neurofibromas plexiformes. Los primeros se originan a partir de ramas nerviosas terminales de la piel y rara vez se desarrollan antes de la pubertad, mientras que los NFP suelen crecer a lo largo de nervios y plexos grandes de cualquier parte del cuerpo, estando presentes desde el nacimiento.
Los NFP se manifiestan de forma muy variable, haciéndose evidentes durante la adolescencia tardía y la adultez temprana, y se diferencian de los neurofibromas localizados (“nodulares” o “atípicos”) en que tienen potencial de transformación maligna; algunos autores los consideran premalignos.
Los NFP pueden tener formas complejas y en ocasiones crecen1 años hasta alcanzar un tamaño muy grande, de incluso el 20% del peso corporal2. Su localización más común es alrededor de la órbita ocular, la cara, las extremidades superiores e inferiores, la espalda, el tórax, el abdomen, el cuello, el plexo braquial y/o el plexo lumbosacro. Presentes en número de uno o varios, no todos tienen que ser sintomáticos (los que provocan dolor y disfunción motora suelen ser los más grandes), pero pueden provocar un impacto clínico importante en las actividades de la vida diaria por asociarse a otros signos y síntomas de distinta gravedad como dolor (a pesar de la analgesia), disfunción neurológica y motora (debilidad y movilidad reducida), compromiso de las vías respiratorias, discapacidad visual o desfiguración. La resolución espontánea de estas manifestaciones es extremadamente improbable. En algunos casos, la enfermedad también se relaciona con dificultades de aprendizaje por los pacientes.
El Instituto Nacional de Salud (NIH) de EE.UU. definió en 1987 los criterios diagnósticos de consenso de la NF1, en base a criterios clínicos (pues los test genéticos no se realizan de rutina). Así, para confirmar un diagnóstico de NF1 deben cumplirse 2 o más de las siguientes características clínicas: a) 6 o más manchas café con leche (diámetro de ≥ 0,5 cm en pacientes prepúberes o ≥ 1,5 cm después de la adolescencia); b) 2 o más neurofibromas o 1 neurofibroma plexiforme; c) pecas en las axilas o ingles; d) glioma óptico; e) 2 o más nódulos de Lisch (hamartomas del iris); f) una lesión ósea específica, como es la displasia del hueso esfenoides o adelgazamiento de la corteza de los huesos largos; y g) un pariente de primer grado con NF1, diagnosticado según estos mismos criterios.
En los casos más graves de la enfermedad, los NFP pueden provocar complicaciones potencialmente mortales debido a la compresión de estructuras vitales, como grandes vasos o la médula espinal, o la obstrucción de las vías respiratorias. Pese a que el pronóstico de la NF1 no es demasiado malo, la enfermedad se asocia con una reducción de 8 a 15 años en la esperanza de vida promedio tanto en hombres como en mujeres, debido principalmente a neoplasias malignas y comorbilidades cardiovasculares. La mortalidad es mayor en pacientes con NFP sintomáticos3, en quienes la causa más común de fallecimiento es la presencia de TMVNP; otras causas de muerte pueden ser shock hipovolémico o insuficiencia respiratoria.
Hasta ahora, las únicas opciones disponibles en el abordaje de pacientes con NF1 son el control farmacológico del dolor y la resección quirúrgica de la mayor cantidad posible de neurofibromas plexiformes. Los problemas neurológicos, ortopédicos y de las vías respiratorias son los principales motivos que conducen a la cirugía. Sin embargo, la extirpación de los NFP no está exenta de dificultades y potenciales complicaciones (alto riesgo de lesión iatrogénica de nervios relacionados y tejidos blandos circundantes, y de hemorragia), y no es una opción para muchos pacientes dado el alto grado de invasividad o proximidad a órganos vitales de la mayoría de los NFP. Cuando sí se alcanza la resección subtotal o parcial de los NFP, casi 1 de cada 5 pacientes experimenta complicaciones quirúrgicas permanentes, incluidas anomalías del habla, parálisis nerviosas y dolor, y más de la mitad (55%) tiene un nuevo crecimiento de la lesión, sobre todo cuando los NFP se resecan de forma incompleta (EMA, 2021).
Acción y mecanismo
Selumetinib es un nuevo inhibidor oral, selectivo y no competitivo con ATP, de las proteínas cinasa 1 y 2 activadas por mitógenos (MEK1/2), que inhibe la proliferación y supervivencia anormales de determinadas células tumorales, como las que forman parte de los neurofibromas. En base a ello, el medicamento, designado como huérfano, ha recibido la autorización condicional para el tratamiento de pacientes pediátricos de 3 años en adelante con neurofibromatosis de tipo 1 (NF1) que presenten neurofibromas plexiformes sintomáticos e inoperables.
Las cinasas MEK1 y MEK2 son componentes críticos de la ruta de señalización bioquímica RAF-MEK-ERK regulada por RAS, la cual se activa con frecuencia en los cánceres humanos por una variedad de tipos de receptores (entre los que se incluyen receptores tirosina cinasas, receptores acoplados a proteína G y receptores de citocinas), estimulando la proliferación de las células tumorales y su supervivencia. Más concretamente, MEK1/2 son dos cinasas con especificidad dual que activan la cinasa regulada por señales extracelulares (ERK) 1/2 a través de la fosforilación de los residuos conservados de treonina y tirosina en su bucle de activación; muestran una alta homología de aminoácidos entre sí (79%) y tienen una capacidad similar para fosforilar y activar sus sustratos ERK1/24. Se comprende, pues, que la actividad inhibitoria de selumetinib sobre las enzimas MEK1/2 detiene la señalización por la vía RAF-MEK-ERK, lo que puede traducirse en un efecto antiproliferativo de células tumorales en que se sobreactiva dicha vía, como las de los neurofibromas (deficitarias de la actividad inhibitoria de neurofibromina-1 sobre RAS).
Los estudios in vitro durante su desarrollo preclínico han demostrado que selumetinib, inicialmente considerado como un potencial candidato en el tratamiento de varios tipos de cáncer, es un inhibidor potente y selectivo de las cinasas MEK1/2 humanas, con valores de CI50 en el rango nanomolar (10-14 nM), siendo inactivo –o mínimamente activo– frente a otras muchas cinasas ensayadas, ni tampoco altera la señalización por las vías de ERK5, mTOR, JNK o p38, lo que puede relacionarse con un buen perfil de seguridad. Investigaciones in vivo en modelos animales con xenoinjertos de varios tipos de tumores humanos demostraron que a dosis clínicamente relevantes la máxima inhibición de la fosforilación de ERK por selumetinib está entre el 40% y el 80%. En dos estudios específicos con un modelo murino de neurofibromatosis tipo 1, esos niveles de dosis similares a las clínicamente relevantes en humanos indujeron una reducción en el número (hasta el 75%) y en el tamaño (hasta el 41%) de los neurofibromas (EMA, 2021).
Aspectos moleculares
Selumetinib (Figura 2) es un fármaco de administración por vía oral que tiene por nombre químico el de 5-[(4-bromo-2-clorofenil)amino]-4-fluoro-6-[(2-hidroxietoxi)carbamoil]-1-metil-1H-benzimidazol-3-io y se presenta en forma de sal de sulfato, correspondiéndose con la fórmula C₁₇H₁₇BrClFN₄O₇S y una masa molecular de 555,8 g/mol (457,7 g/mol en su forma de base). Se trata de una molécula pequeña que no exhibe estereoisomería y se presenta como un polvo cristalino no higroscópico, de color blanco a amarillo, con baja solubilidad (prácticamente insoluble en agua y solo ligeramente soluble en etanol y acetonitrilo).
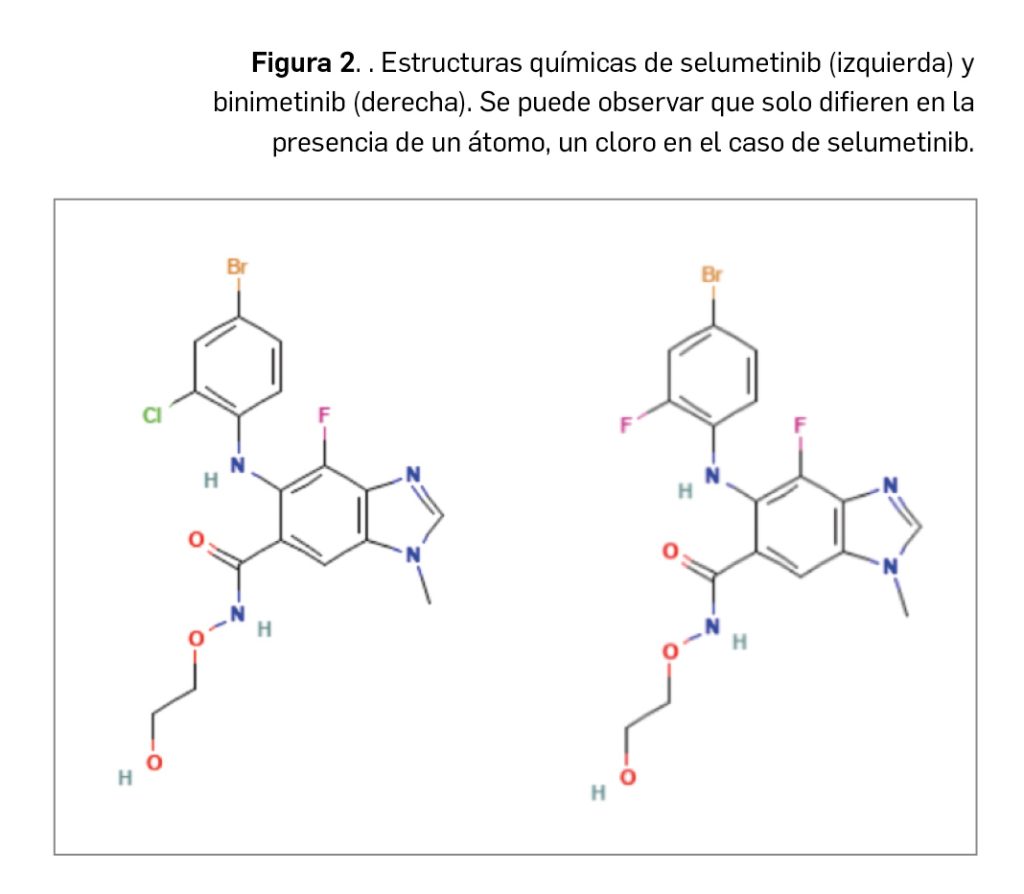
Selumetinib guarda una muy estrecha relación con binimetinib, otro inhibidor selectivo de tirosina cinasas MEK1/2 comercializado en España en 2019 para el tratamiento en combinación con encorafenib de pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600. Como binimetinib, el nuevo fármaco está estrechamente relacionado estructural y farmacológicamente con otros miembros de la serie de inhibidores de proteína cinasas, en la que se encuadran un gran número de principios activos comercializados en España, resultantes de la optimización funcional mediante modelización molecular a partir de una serie de 2-fenilaminopirimidinas, de donde surgió el imatinib, cabeza de serie del grupo. Aunque se aprecia una diversidad estructural importante en el grupo, todos presentan heterociclos derivados azólicos y guardan –en mayor o menor grado– una familiaridad química con la molécula de ATP (o, en su caso, con la de GTP, como sucede en las cinasas MAPK), con la cual algunos de los principios activos compiten para provocar el bloqueo de la cinasa correspondiente. Se han desarrollado modelos moleculares de relación estructura-actividad para este grupo de sustancias y, en todos los casos, las interacciones estéricas y electrostáticas han demostrado ser determinantes para el efecto inhibitorio sobre la tirosina cinasa.
Eficacia y seguridad clínicas
La eficacia y seguridad de selumetinib en su indicación y pauta aprobadas (hasta 50 mg dos veces al día, con el estómago vacío) han sido evaluadas en un único ensayo de fase 1/2 de diseño abierto, multicéntrico y de un solo grupo (estudio SPRINT), aún en marcha. Los datos de eficacia inicialmente divulgados (Gross et al., 2020) se refieren a 50 niños de entre 3 y 18 años con neurofibromatosis tipo 1 enrolados en el estrato 1 de la fase 2 del ensayo, quienes debían presentar neurofibromas plexiformes inoperables5 relacionados con una morbilidad importante. Los pacientes fueron tratados de forma continua –en ciclos de 28 días y hasta pérdida de beneficio clínico– con una dosis del fármaco de 25 mg/m2.
La mediana de edad de los pacientes al inicio fue de 10,2 años, el 60% eran varones y el 84% de raza blanca. Desde el punto de vista clínico, la mediana del volumen del NFP diana (el que causaba síntomas o complicaciones clínicas importantes) al inicio del ensayo fue de 488 ml, el cual se asociaba en más del 20% de los casos a desfiguración, discapacidad motora, dolor, afectación de las vías respiratorias, disfunción visual y vesical/intestinal, entre otras. Se excluyeron del estudio aquellos candidatos que presentaran determinadas toxicidades oculares previas.
La variable principal de eficacia fue la tasa de respuesta objetiva (en adelante, TRO), definida como la suma de respuestas completas –desaparición del NFP diana– y de respuestas parciales –reducción ≥ 20% del volumen del NFP tras 3-6 meses en comparación con el valor basal–. Para ello, se hicieron análisis volumétricos del NFP por resonancia magnética cada 4 meses los primeros 2 años (después, cada 6 meses) y se consideró la respuesta según los criterios validados REiNS6, midiéndose también otros resultados clínicos, como pruebas funcionales y resultados comunicados por los pacientes. La respuesta debía reconfirmarse tras al menos 3 meses.
Los resultados más actualizados, basados en un corte de datos de marzo de 2021 (AEMPS, 2022) reflejan una tasa de respuesta objetiva del 68% (IC95% 53,5-80,5). Si bien los 34 pacientes respondedores tuvieron una respuesta parcial (en ningún caso individual fue completa), es preciso subrayar que esas cifras son superiores a las estimadas por una revisión centralizada independiente según criterios REiNS, que anteriormente (con fecha de corte de datos de 2018) había apuntado a una TRO del 44%. Tras un seguimiento mediano superior a 41 meses, no se alcanzó la mediana de duración de la respuesta, pero se constató que esa duración era superior a 24 y 36 meses en el 76% (26/34) y el 62% (21/34) de los pacientes respondedores, respectivamente.
Entre otras variables secundarias, destaca la mediana del tiempo hasta la aparición de la respuesta, que fue de 7,2 meses (intervalo: 3-38 meses), mientras que la mediana de tiempo hasta la reducción máxima del volumen del NFP fue de 15,1 meses desde el inicio (intervalo: 3-62 meses). En cambio, no se alcanzó la mediana del tiempo transcurrido desde el inicio del tratamiento hasta la progresión de la enfermedad durante el tratamiento (supervivencia libre de progresión, SLP). Con respecto a los resultados informados por los pacientes pediátricos, tras 1 año de tratamiento la disminución media en las puntuaciones de intensidad del dolor auto-reportada –escala NRS– fue de -2,1 puntos, lo que se considera una mejora clínicamente significativa, como también lo fue la mejora en la interferencia del dolor en la vida diaria informada por los niños y los padres (38% y 50%, respectivamente), en la calidad de vida relacionada con la salud (48% y 58%, respectivamente), y en los resultados funcionales de fuerza (56%) y rango de movilidad (38%) (Gross et al., 2020).
Más recientemente se han publicado datos de un estudio de soporte de fase 2 en el que participaron pacientes pediátricos asintomáticos con NF1 y presencia de NFP no operables que podrían potencialmente provocar síntomas, pero que en el momento de inclusión en el estudio no suponían ninguna comorbilidad significativa. El tratamiento oral diario con selumetinib indujo una respuesta parcial confirmada en el 72% de los pacientes tras una media de 41 ciclos de tratamiento (cada uno de 28 días). Durante ese periodo, la mayoría de las funciones orgánicas evaluadas al inicio, como la capacidad visual, motora, vesical/intestinal o la funcionalidad de las vías aéreas, se mantuvieron en los límites normales sin empeoramiento clínicamente significativo, indicativo de que no se desarrolló ninguna manifestación relacionada con los NFP (Gross et al., 2022).
Desde el punto de vista de la seguridad, los escasos datos disponibles proceden de 74 pacientes que recibieron selumetinib a distintas dosis durante las dos fases del ensayo SPRINT, que están en línea con el perfil toxicológico observado en adultos con varios tipos de tumores en otros estudios (n= 347, en hasta 7 estudios clínicos). Con una mediana de exposición al fármaco de 55 meses (61% tratados durante > 4 años), el tratamiento fue mejor tolerado entre los niños con NF1 de 12 a 18 años en comparación con los más pequeños. Independientemente de su gravedad, las reacciones adversas más frecuentes entre los que recibieron la pauta aprobada de selumetinib (≥ 60%) fueron: vómitos (86%), diarrea (81%), aumento de niveles sanguíneos de creatinfosfocinasa (77%), náuseas (77%), piel seca (65%), pirexia (61%) y dermatitis acneiforme (61%).
Aunque la toxicidad a nivel gastrointestinal y cutáneo constituye el aspecto más relevante del perfil de seguridad, la fatiga, infecciones de uñas, estomatitis, erupciones no acneiformes y alteraciones en la analítica sanguínea (hemoglobina y albúmina reducidas y elevación de transaminasas) también tuvieron una notable incidencia, afectando a más de la mitad de los pacientes. En consecuencia, se notificó una alta tasa de interrupción (82%) y reducciones de la dosis (39%) por eventos adversos, sobre todo vómitos (32%) y náuseas (19%), infecciones de uñas (23%), diarrea (15%) y pirexia (11%); no obstante, solo un 12% tuvieron que discontinuar el tratamiento como consecuencia de efectos tóxicos posiblemente relacionados con selumetinib. La mayoría de las reacciones adversas descritas fueron leves-moderadas en severidad, destacando entre los casos de eventos graves (grado 3) las siguientes: diarrea (3%), anemia (3%), pirexia (3%), creatinfosfocinasa elevada en sangre (3%), edema periférico (1%) y vómitos (1%).
Aspectos innovadores
Selumetinib es un nuevo inhibidor oral, selectivo y no competitivo con ATP, de las proteínas cinasa 1 y 2 activadas por mitógenos (MEK1/2), componentes críticos de la ruta de señalización bioquímica RAF-MEK-ERK regulada por RAS, la cual se activa con frecuencia en los cánceres humanos por una variedad de tipos de receptores y estimula la proliferación de las células tumorales y su supervivencia. Se comprende, pues, que la actividad inhibitoria de selumetinib detiene la señalización por dicha vía, lo que se traduce en un efecto antiproliferativo en células tumorales como las de los neurofibromas (deficitarias de la actividad inhibitoria sobre RAS de neurofibromina-1). En base a ello, el medicamento7, designado como huérfano, ha recibido una autorización condicional de comercialización para el tratamiento de pacientes pediátricos de 3 años en adelante con neurofibromatosis de tipo 1 (NF1) que presenten neurofibromas plexiformes (NFP) sintomáticos e inoperables.
Los datos –limitados– de eficacia que han sustentado esa autorización condicional derivan de la fase 2 del estudio SPRINT, con diseño abierto y de un solo grupo, en que la administración oral diaria de selumetinib hasta pérdida de beneficio clínico en niños (n= 50, mediana de 10 años de edad) con NF1 y presencia de NFP inoperables y asociados a una morbilidad importante determinó una tasa de respuesta objetiva del 68%, en todos los casos respuestas parciales (reducción ≥ 20% del volumen del NFP diana tras 3-6 meses, confirmado por resonancia); la mediana del cambio óptimo respecto al inicio fue de -28% del tamaño del NFP y ninguno de los pacientes tratados experimentó progresión de la enfermedad. Tras un seguimiento superior a 41 meses, no se alcanzó la mediana de duración de la respuesta, que en casi 2 de cada 3 pacientes respondedores (62%) perduró más allá de los 3 años, sin poderse establecer la mediana de tiempo hasta progresión de la enfermedad (SLP). La mediana del tiempo hasta aparición de la respuesta estuvo en torno a los 7 meses, y la reducción del volumen del NFP fue máxima tras 15 meses desde el inicio, verificándose con independencia del estatus del neurofibroma al inicio.
Esos resultados se complementaron con los autoinformados por los pacientes y/o sus padres8, tales como una mejora clínicamente significativa de la intensidad del dolor en aproximadamente un cuarto de los pacientes, o una tendencia a la mejora de la interferencia del dolor en la vida diaria, de las capacidades funcionales y de la calidad de vida relacionada con la salud en algunos pacientes, pero sin correlación a nivel poblacional entre la reducción en el tamaño del tumor y la mejoría de un determinado parámetro clínico. Además, en un estudio de soporte de fase 2 en pacientes pediátricos asintomáticos con NFP inoperables con potencial de provocar síntomas, selumetinib indujo una respuesta parcial confirmada en el 72% de los pacientes, tras una media de > 38 meses de tratamiento, lo que se relacionó con ausencia de manifestaciones clínicas por los NFP.
Por otro lado, su perfil toxicológico –caracterizado también por datos limitados–, aunque importante, parece aceptable y clínicamente manejable con medidas de soporte y ajustes posológicos, en línea con lo conocido para otros inhibidores de MEK-2, como binimetinib o trametinib. Los principales signos de seguridad asociados a selumetinib son la toxicidad gastrointestinal (vómitos, diarrea y náuseas) y dermatológica (piel seca y dermatitis acneiforme), que afectan a una mayoría de pacientes (> 60%), pero con menor incidencia a mayor edad de los niños. La fiebre, la fatiga, las infecciones de uñas o las alteraciones en la analítica sanguínea son también frecuentes, reportadas en más de la mitad de los pacientes. En general se trata de reacciones adversas leves-moderadas que, aunque requieren frecuentes interrupciones o reducciones de la pauta posológica, solo suponen una tasa del 12% de discontinuación del tratamiento (sobre todo, por problemas digestivos). Entre las graves, destacan (3%) la diarrea, anemia, fiebre y aumento de creatinfosfocinasa en sangre.
En el contexto de una enfermedad neoplásica rara, crónica y severamente debilitante para la que no existe ninguna opción farmacológica aprobada (ni para curar, prevenir o manejar específicamente la sintomatología), se comprende que la inclusión de un control con placebo no se considere ético. Por ello, a pesar de las diferencias metodológicas entre estudios que limitan la robustez de las conclusiones, el EPAR de la EMA recoge la comparativa –meramente descriptiva– de los datos del citado estudio con dos controles externos: a) pacientes incluidos en el brazo placebo del estudio aleatorizado de tipifarnib en niños y adultos con NF1 y NFP progresivos, y b) datos de la historia natural de la enfermedad a partir de un estudio observacional sin intervención. Por ejemplo, en estos últimos, ningún paciente mostró un cambio > 20% en el volumen de los NFP en periodos de 1 año. En consecuencia, dado que la evolución sin tratamiento se relaciona con un aumento o, en el mejor de los casos, estabilidad del tamaño de los NFP a lo largo del tiempo, la respuesta de larga duración observada en el estudio clínico (reducción del volumen de los NFP) puede ser atribuida a selumetinib y probablemente se asociará con una menor sintomatología y morbilidad; no obstante, por el limitado seguimiento de los pacientes (aunque superior a 3 años) aún se desconoce la magnitud real del efecto sobre la supervivencia libre de progresión.
En definitiva, con un mecanismo de acción novedoso en su indicación (ya establecido en otras patologías oncológicas), parece probada la eficacia de administrar selumetinib en infancia y adolescencia (los periodos de mayor riesgo de progresión de la NF1), probablemente relacionada con la mejora en los resultados clínicos individuales, tales como la calidad de vida, la intensidad de dolor o las capacidades motoras; una eficacia que podrá ser variable en los distintos pacientes dada la heterogeneidad de las manifestaciones. Se desconoce si incidirá sobre el curso natural de la enfermedad o afectará al desarrollo de los niños, y también hay incertidumbres sobre su seguridad a largo plazo y la evolución de los tumores si se discontinúa el tratamiento, pero se espera que los estudios comparativos y mejor diseñados ya en marcha (en pacientes pediátricos de incluso menos de 3 años y en pacientes adultos) aporten una evidencia más robusta.
Hay que considerar que los neurofibromas son tumores benignos que pueden llegar a afectar a la mitad de los pacientes con la enfermedad, en quienes hasta ahora la cirugía, con sus complicaciones, era la única opción terapéutica. Esa mitad de pacientes será la que se beneficie potencialmente de selumetinib como el único tratamiento disponible frente a los NFP inoperables que general gran morbilidad: si bien no será un tratamiento erradicador, podrá facilitar una cirugía posterior.

Bibliografía
-
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Ficha técnica de Koselugo® (selumetinib). 2022. Disponible en: https://www.ema.europa.eu/en/documents/product-information/koselugo-epar-product-information_es.pdf.
-
- Asociación de Afectados por la Neurofibromatosis. ¿Qué es la neurofibromatosis? Disponible en: https://neurofibromatosis.es/que-es-neurofibromatosis/ (consultado a 19 de septiembre
de 2023).
- Asociación de Afectados por la Neurofibromatosis. ¿Qué es la neurofibromatosis? Disponible en: https://neurofibromatosis.es/que-es-neurofibromatosis/ (consultado a 19 de septiembre
-
- European Medicines Agency (EMA). Koselugo®. European Public Assessment Report (EPAR). 2021. EMA/549867/2021. Disponible en: https://www.ema.europa.eu/en/documents/assessment-report/koselugo-epar-public-assessment-report_en.pdf.
-
- Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med. 2020; 382(15): 1430-42. DOI: 10.1056/NEJMoa1912735. Erratum in: N Engl J Med. 2020; 383(13): 1290.
-
- Gross AM, Glassberg B, Wolters PL, Dombi E, Baldwin A, Fisher MJ et al. Selumetinib in children with neurofibromatosis type 1 and asymptomatic inoperable plexiform neurofibroma at risk for developing tumor-related morbidity. Neuro Oncol. 2022; 24(11): 1978-88. DOI: 10.1093/neuonc/noac109.
1 A medida que los NFP tienen una mayor velocidad de crecimiento, requieren un mayor uso de analgesia farmacológica.
2 Se ha observado que los NFP crecen más rápidamente durante la primera década de la vida y, si bien la tasa de crecimiento es muy variable entre los pacientes (oscila entre el +3%/año hasta el +16%/año), en niños más pequeños es generalmente mucho mayor que en adolescentes o adultos, llegando a superar la tasa prevista según el aumento de su peso corporal o su índice de masa corporal. También se ha visto un crecimiento más lento conforme más grande es el neurofibroma. En ocasiones, se han descrito contracciones espontáneas de hasta un tercio de las lesiones, a una velocidad de cambio medio cercana al -3%/año, nunca superior a -20%/año.
3 Un análisis retrospectivo de datos de los registros clínicos de niños con NF1 reportó una mayor tasa de mortalidad en niños con NFP sintomáticos (3,2%) frente a aquellos sin NFP o con NFP asintomáticos (0,5%).
4 En contraste con la especificidad de sustrato exclusiva de MEK1/2 para ERK1/2, se han identificado cientos de proteínas como sustratos de ERK, lo que explica cómo la vía RAF-MEK-ERK puede influir en muchos procesos celulares, incluida la proliferación y supervivencia de células tumorales.
5 Se definió un NFP como “no operable” cuando era imposible extirparlo quirúrgicamente por completo sin riesgo de morbilidad considerable, debido al atrapamiento o la proximidad a estructuras vitales, la invasividad o la vascularización elevada.
6 La Colaboración Internacional de Evaluación de Respuesta en Neurofibromatosis y Schwannomatosis (REiNS) recomienda los siguientes instrumentos de resultados informados por el paciente para evaluar el dolor y la funcionalidad física en ensayos clínicos de NF1: a) la escala de calificación numérica-11 (NRS-11) para la evaluación (autoinformada) de la intensidad del dolor en pacientes de ≥ 8 años; b) el índice de interferencia del dolor, autoinformado en pacientes de 6 a 24 años e informado por los padres en pacientes de 6 a 18 años; y c) el sistema de información de medición de resultados informados por el paciente (PROMIS) para medir la movilidad y la función de las extremidades superiores, autoinformado en pacientes de 8 a 17 años e informado por los padres entre 5 y 17 años.
7 El medicamento está formulado en cápsulas duras, que no se consideran apropiadas para pacientes muy jóvenes por su tamaño, al incrementar las dificultades de su toma oral. El laboratorio se ha comprometido con las agencias reguladoras a desarrollar una forma farmacéutica adecuada a la población pediátrica.
8 El diseño abierto del estudio limita la representatividad de estas variables, pues el conocimiento por parte de los pacientes y sus padres del tratamiento recibido puede llevar a una sobreestimación de su efecto.
