Metabolopatías y su tratamiento
 Nº433
Nº433

Se definen como metabolopatías aquellas patologías en las que se produce la alteración de una o más vías bioquímicas que forman parte de algún proceso fisiológico normal. La clasificación más habitual es aquella basada en los efectos celulares, existiendo:
- metabolopatías que provocan la acumulación de sustratos fisiológicos, como consecuencia de un defecto en el anabolismo o catabolismo de moléculas complejas;
- metabolopatías que causan intoxicación por metabolitos tóxicos, derivadas de su acumulación;
- y metabolopatías que causan déficit de energía, limitando la disponibilidad de energía en el organismo.
Las estrategias terapéuticas para el abordaje de las metabolopatías congénitas se basan principalmente en compensar los efectos de la vía metabólica afectada. Entre estas estrategias se encuentran:
- reducir el catabolismo;
- limitar la ingesta de alimentos u otros productos que permitan o faciliten la producción de la sustancia tóxica;
- incrementar la excreción de metabolitos tóxicos;
- potenciar la actividad enzimática residual;
- o el uso de terapias avanzadas (terapia génica, terapia celular somática, ingeniería tisular).
En líneas generales, las metabolopatías se caracterizan por ser enfermedades de muy baja prevalencia, llamadas enfermedades raras. Existen dos definiciones de enfermedad rara: i) en la Unión Europea: aquella que tiene una prevalencia inferior a 5 casos por 10.000 habitantes (1:2.000); ii) en Estados Unidos: aquella que afecta a menos de 200.000 personas en ese país (1:1200). Muchos de los medicamentos utilizados en el tratamiento de estas patologías carecen de otras indicaciones y tienen una demanda muy baja: son los conocidos como medicamentos huérfanos, que se definen como aquellos medicamentos destinados a la prevención, diagnóstico o tratamiento de enfermedades raras o de enfermedades graves más comunes pero que difícilmente serían comercializados por falta de perspectivas de venta una vez en el mercado.
Metabolopatías de las bases nucleicas
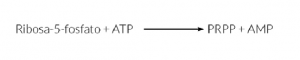
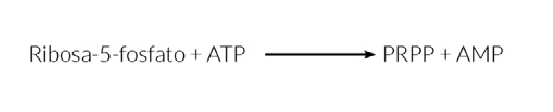
La biosíntesis de nucleótidos de purinas y pirimidinas precisa del fosforribosilpirofosfato (PRPP), generado por acción de la PRPP sintetasa, y requiere de energía en forma de ATP:
Metabolopatías congénitas de bases purínicas
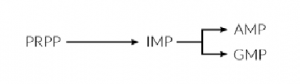
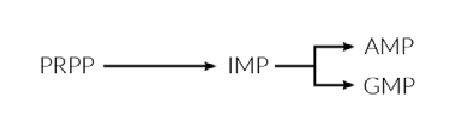
La biosíntesis de nucleótidos purínicos (adenina, guanina) comienza con PRPP y conduce al primer nucleótido purínico completo, la inosina-5´-monofosfato (IMP), que constituye el punto central del proceso, ya que puede ser convertido tanto en AMP como en GMP, a través de dos rutas metabólicas diferentes.
El producto final del catabolismo de los nucleótidos purínicos es el ácido úrico, un producto de naturaleza muy poco soluble que es excretado con la orina bajo la forma de cristales de urato monosódico. Una de las enzimas que interviene en este proceso es la xantina oxidasa, diana de los principales fármacos utilizados en el manejo terapéutico de la hiperuricemia.
El depósito de los cristales de urato monosódico en el fluido sinovial y las articulaciones, provoca un cuadro clínico de inflamación intensa conocido como gota (Figura 1), que se trata, en la actualidad, con antiinflamatorios no esteroideos (AINE) como el ibuprofeno, el naproxeno o la indometacina. En pacientes no respondendores a esos fármacos o en aquellos cuya administración esté contraindicada, pueden emplearse glucocorticoides y colchicina. Esta última posee un margen terapéutico estrecho, motivo por el cual ha sido desplazada por los AINE, con un margen de seguridad mayor.
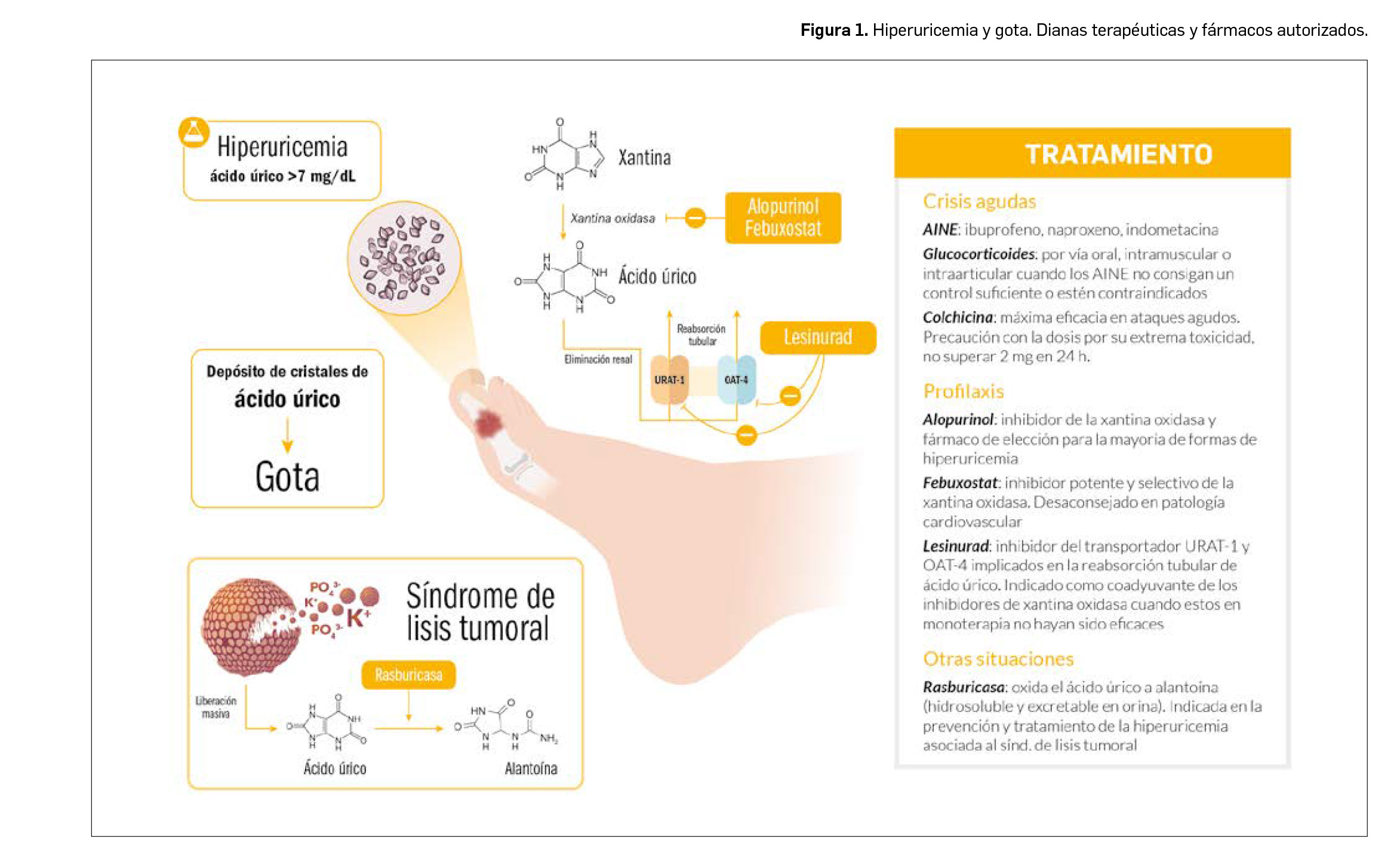
Para el tratamiento de la hiperuricemia más o menos crónica pueden emplearse los inhibidores de la xantina oxidasa como alopurinol (purínico) o febuxostat (no purínico), que podrían incrementar el riesgo de sufrir crisis de gota al inicio de tratamiento, al producir la movilización de los depósitos tisulares de ácido úrico, por lo que se aconseja la profilaxis conjunta con un AINE o colchicina durante los primeros meses de tratamiento. La administración de febuxostat se ha relacionado con una mayor incidencia de efectos adversos cardiovasculares graves en comparación con alopurinol, por lo que deberán extremarse las precauciones en pacientes con cardiopatía isquémica y/o insuficiencia cardiaca.
Con un mecanismo de acción diferente, lesinurad actúa impidiendo la reabsorción tubular de ácido úrico, mediante la inhibición del transportador URAT-1. El principal efecto adverso limitante de su uso es la nefrotoxicidad por lo que, en caso de elevación notable de la creatinina sérica, se debe suspender inmediatamente el tratamiento.
La rasburicasa constituye la enzima urato-oxidasa de origen recombinante, obtenida a partir de una cepa de Saccharomyces cerevisiae, que actúa como agente uricolítico catalizando la oxidación enzimática del ácido úrico a alantoína, un producto hidrosoluble, que se excreta fácilmente por vía renal. Está indicado en infusión intravenosa para el tratamiento y profilaxis de la hiperuricemia aguda en pacientes con neoplasia hematológica maligna con elevada carga tumoral y riesgo de lisis celular.
Entre las alteraciones del metabolismo de las purinas también se incluyen el síndrome de Lesch-Nyhan, el síndrome de Kelley-Seegmiller, la xantinuria y el síndrome de inmunodeficiencia severa combinada (SCID), así como otras patologías causadas por déficit de cualquier enzima ligada al metabolismo purínico.
Metabolopatías congénitas de bases primidínicas
La síntesis de pirimidinas es más simple que la de bases purínicas. El punto de partida es el carbamil-fosfato, que se condensa con el aspartato para generar ácido orótico u orotato, y que, gracias a la acción de la enzima bifuncional UMP sintasa, generará el nucleótido uridilato (UMP):

La degradación de los nucleótidos pirimidínicos se inicia a partir de los correspondientes nucleótidos monofosfato. El UMP y el CMP sufren una serie de transformaciones de carácter irreversible hasta β-alanina, mientras que la TMP es transformada finalmente en β-aminoisobutirato.
Las alteraciones enzimáticas del metabolismo de las bases pirimidínicas son pocas y extremadamente infrecuentes, siendo la más característica la aciduria orótica hereditaria, causada por el déficit de la enzima bifuncional UMP sintasa, dando como resultado un aumento en la excreción de ácido orótico (de ahí su nombre).
Metabolopatías de los pigmentos
Biosíntesis del grupo hemo: porfirias
La inactividad parcial o completa de una o varias enzimas participantes en la síntesis del grupo hemo origina un cuadro clínico conocido como porfiria (Figura 2), caracterizado, fundamentalmente, por la aparición de neuropatía, lesiones cutáneas, o la asociación de ambas.

Los factores implicados que con mayor frecuencia pueden desencadenar una crisis aguda de porfiria son el tabaco, el consumo de alcohol, la administración de determinados fármacos, la presencia de infecciones, el estrés y la exposición solar.
El tratamiento de la porfiria aguda consistirá en la eliminación de los factores desencadenantes, seguido de la administración de grandes cantidades de glucosa (400-500 g/día), debido al efecto inhibidor de esta sobre la ALA sintasa. Adicionalmente, la sintomatología asociada al cuadro puede abordarse mediante la administración de analgésicos opiodes (dolor abdominal intenso), β-bloqueantes (hipertensión), suero salino (hiponatremia) y diazepam o clonazepam (crisis convulsivas).
La fotosensibilidad propia de las porfirias con predominio de afectación cutánea puede paliarse mediante el uso de barreras físicas, y el empleo de filtros solares y β-carotenos, siendo estos últimos capaces de activar la protoporfirina depositada en la piel. Para el caso concreto de la porfiria cutánea tardía, ha demostrado ser eficaz la realización de flebotomías. El arginato de hemina se utiliza en el tratamiento de las porfirias agudas hepáticas.
Recientemente, se ha autorizado en la UE el uso de afamelanotida, el primer medicamento en implante subcutáneo indicado para prevenir la fototoxicidad en pacientes adultos afectados por protoporfiria eritropoyética.
Degradación del grupo hemo: metabolismo de la bilirrubina
La degradación del grupo hemo procedente de la hemoglobina y otras proteínas comienza en el sistema fagocítico mononuclear (SFM) (Figura 3), donde es transformado en biliverdina por acción de la hemooxigenasa, y posteriormente en bilirrubina por la biliverdina reductasa. La bilirrubina accede al plasma, donde es captada por los hepatocitos. Una vez en el retículo endoplasmático liso, sufre un proceso de conjugación con ácido glucurónico por acción de la UDP glucuroniltransferasa, dando lugar a bilirrubina conjugada, que posteriormente será transformada en urobilinógeno y eliminado mayoritariamente por vía fecal.
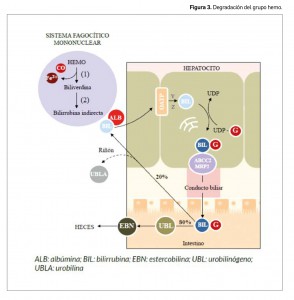
Los errores congénitos que afectan al proceso de conjugación cursan, predominantemente, con elevación de los niveles de bilirrubina no conjugada; En este contexto, se encuentran los síndromes de Gilbert y Crigler-Najjar, producidos por el déficit parcial o completo de la enzima UDP-glucuroniltransferasa, respectivamente. Por el contrario, las alteraciones en la conjugación de la bilirrubina en el hepatocito, elevan los niveles de bilirrubina conjugada, como ocurre en los síndromes de Rotor y Dubin-Johnson, este último asociado a defectos en el transportador ABCC2, compartido por otros aniones orgánicos.
Metabolismo de ácidos biliares
La biosíntesis de ácidos biliares primarios (cólico, quenodesoxicólico) ocurre en el hepatocito a partir del colesterol, en una ruta metabólica catalizada por la 7-α-hidroxilasa. La administración de ácido cólico exógeno ralentiza la producción de ácidos biliares, mediante la activación del receptor farnesoide X, que atenúa la transcripción del gen que codifica para tal enzima, limitante del proceso, resultando útil en esta y otras deficiencias primarias como la producida en la xantomatosis cerebrotendinosa.
Metabolopatías de los aminoácidos
El nitrógeno procedente de la desaminación durante el catabolismo de los aminoácidos se elimina en el hígado a través del ciclo de la urea. Las alteraciones del ciclo de la urea pueden presentarse tanto en el periodo neonatal como en la infancia o la edad adulta, pudiendo conducir a un cuadro prolongado de hiperamonemia (que pueden desencadenarse tras la ingesta de altas cantidades de proteínas, largos periodos de inanición, etc).
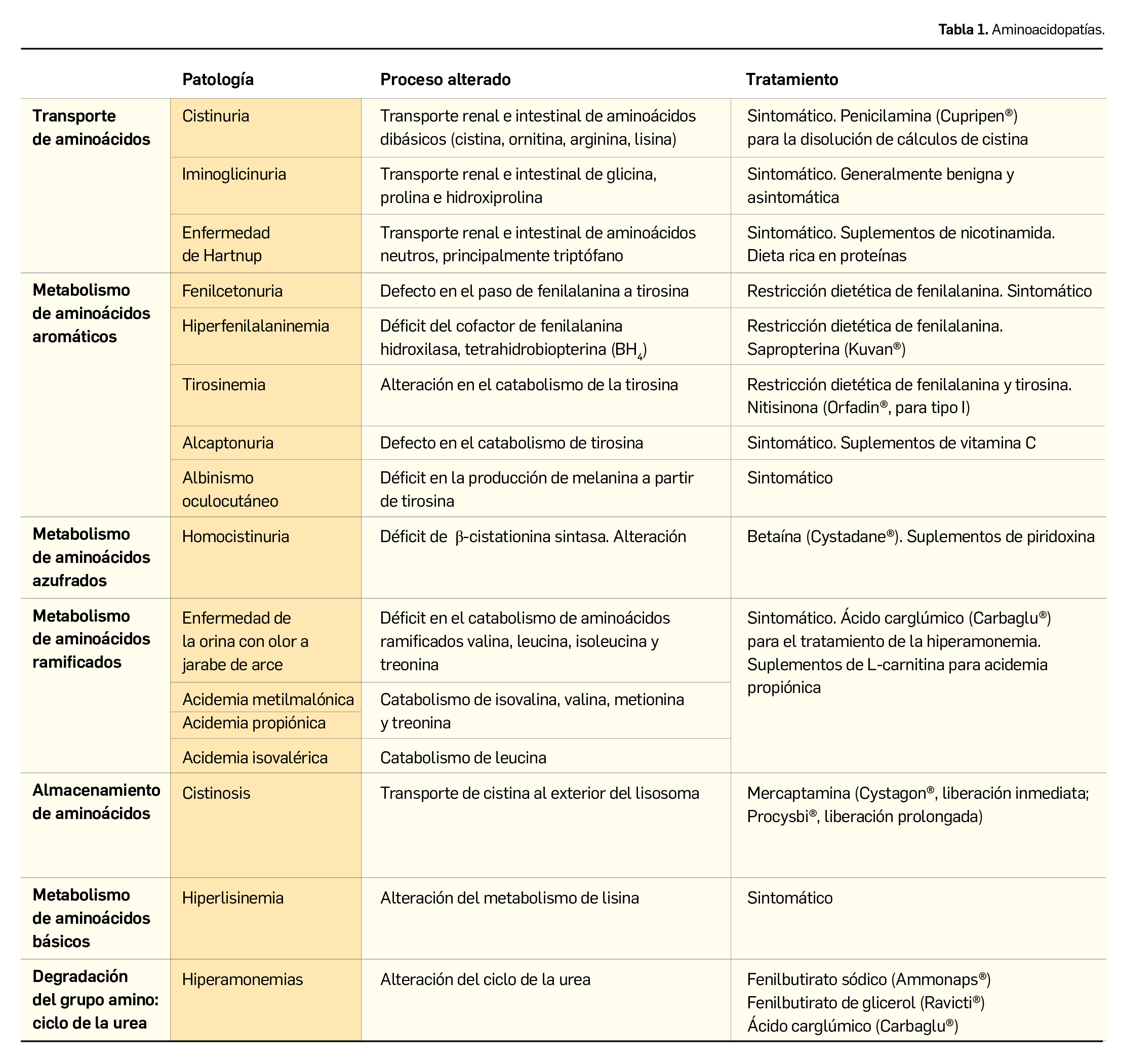
El abordaje terapéutico de tales alteraciones pasa por reducir el aporte de proteínas de la dieta, suplementar con arginina o citrulina en aquellos casos en los que proceda, y proporcionar una vía alternativa de eliminación del nitrógeno metabólico. El fenilbutirato sódico y, más recientemente el fenilbutirato de glicerol, consiguen disminuir los niveles plasmáticos de amoníaco. Específicamente, el ácido carglúmico está indicado en pacientes con hiperamonemia debida a la deficiencia de N-acetilglutamato sintasa. En la Tabla 1 se recogen las principales aminoacidopatías clasificadas según la parte de la ruta metabólica alterada, con indicación del tratamiento recomendado.
Metabolopatías de los glúcidos
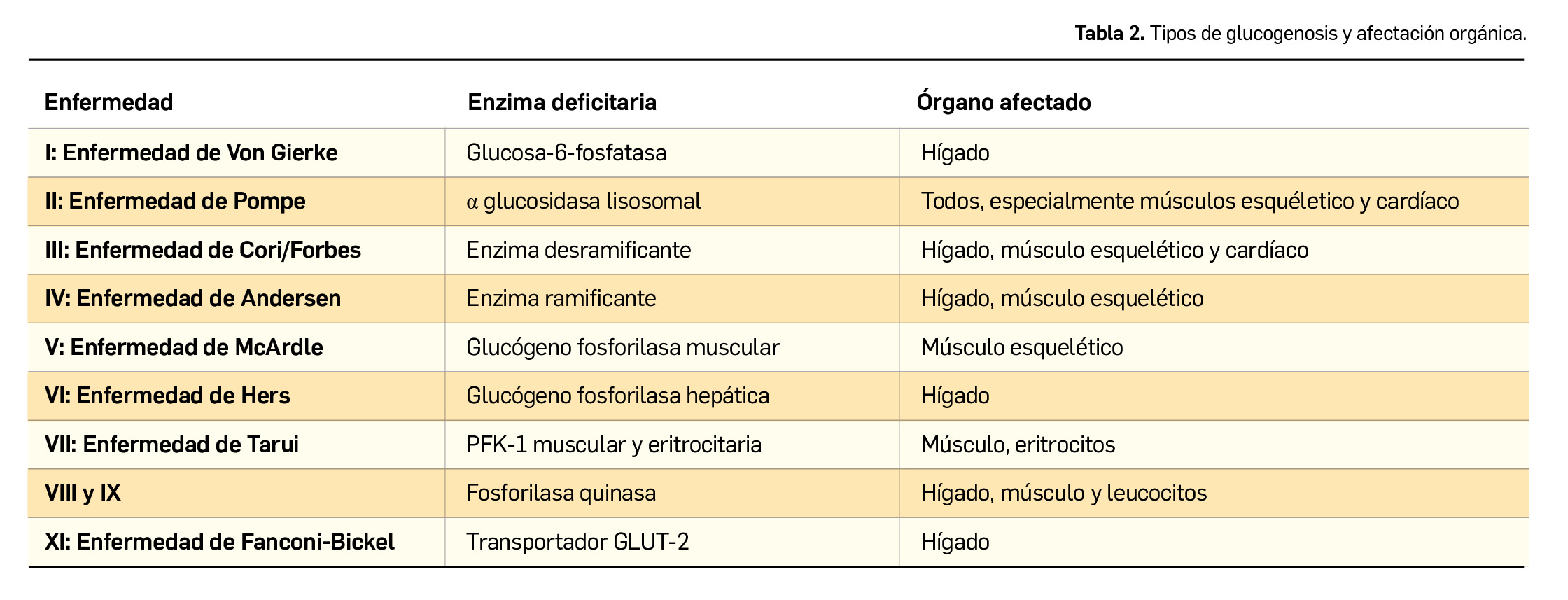
Glucogenosis
Constituyen un grupo de enfermedades del almacenamiento del glucógeno que pueden afectar al hígado, a los músculos o a ambos (Tabla 2). La enfermedad de Pompe, causada por el déficit de la enzima desramificante, es la única frente a la cual se dispone de tratamiento específico, mediante terapia de sustitución enzimática con alglucosidasa α.
Trastornos del metabolismo de monosacáridos
Se incluyen en este grupo los errores del metabolismo de monosacáridos como la galactosa y la fructosa. En la Figura 4 se representa el metabolismo de estos glúcidos.
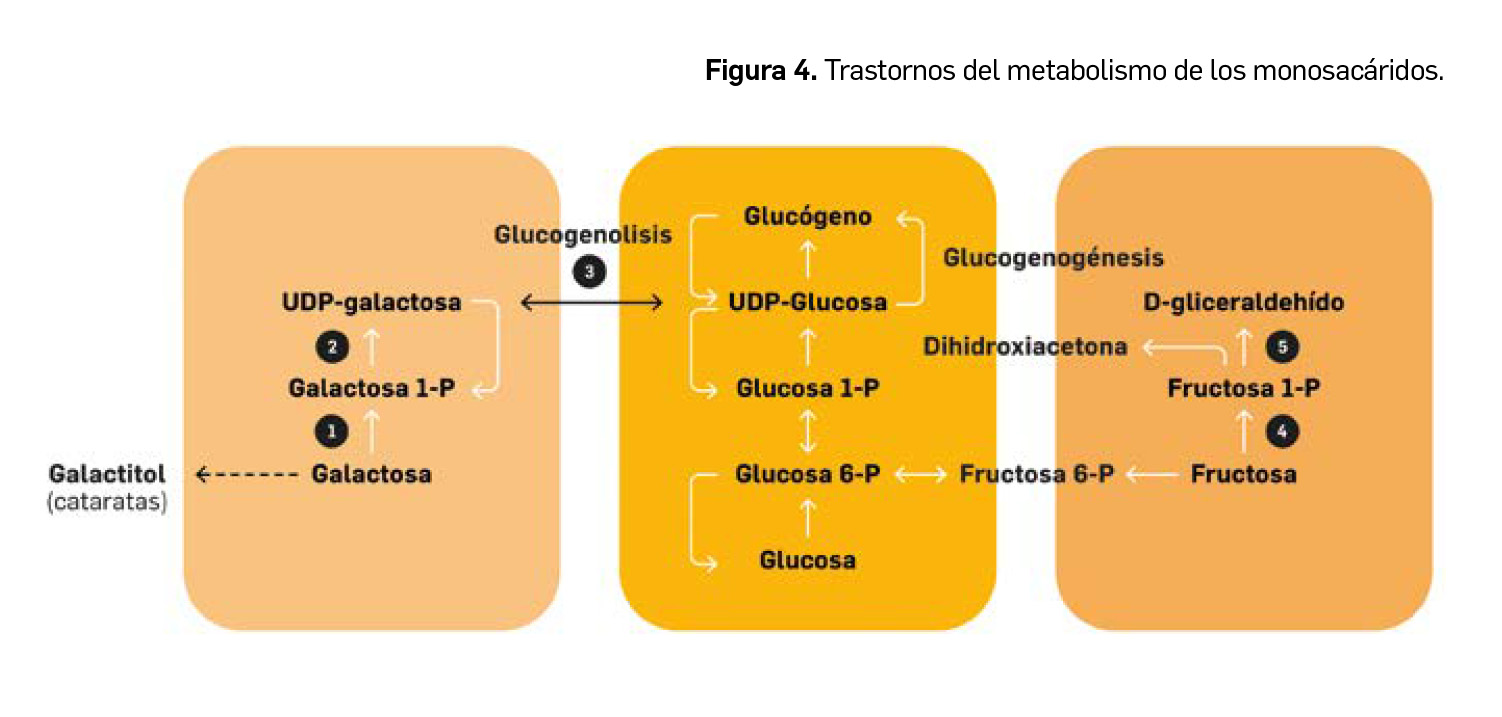
La galactosemia constituye una enfermedad caracterizada por la incapacidad de metabolizar la galactosa a glucosa. Existen tres formas de la enfermedad, producidas por el déficit de cada una de las enzimas participantes en dicha ruta. Así, la deficiencia de galactosa-1-P-uridil transferasa (2) produce la galactosemia clásica, la forma más común y grave de la enfermedad. Como consecuencia del déficit de galactoquinasa (1), una forma mucho menos frecuente de la enfermedad, la galactosa se metaboliza siguiendo una ruta alternativa que culmina con la formación de galactitol, y que se acumula en tejidos como el cristalino, produciendo cataratas. El déficit de galactosa epimerasa (3) incluye una variante que, en su forma grave se asemeja a la clásica.
Por otra parte, la intolerancia hereditaria a la fructosa está provocada por una deficiencia congénita de aldolasa B (5), que provoca una reducción de la glucogenolisis y de la gluconeogénesis.
Déficit de disacaridasas intestinales
Los déficits de disacaridasas intestinales incluyen las deficiencias de una o varias enzimas implicadas en el desdoblamiento de disacáridos en monosacáridos, en el tracto intestinal. La patología más común asociada a este tipo de déficits es la intolerancia a la lactosa, cuya forma más frecuente es la secundaria, generalmente provocada por un daño intestinal temporal asociada a una gastroenteritis vírica. Actualmente, se dispone de complementos nutricionales que contienen lactasa, recomendables para su uso ocasional, y que permiten paliar el déficit de tal enzima en aquellas situaciones en las que se prevea una ingesta abundante de lactosa.
Metabolopatías de orgánulos celulares
Metabolopatías lisosomales
Los lisosomas constituyen los orgánulos celulares encargados del proceso de “digestión celular”. Contienen en su interior enzimas de tipo hidrolasa, de las que se han descrito hasta 40 tipos. Las enzimas hidrolíticas, sintetizadas en el retículo endoplasmático rugoso, en el aparato de Golgi por transporte vesicular sufren una glicosilación terminal de la cual resultan con cadenas glucídicas ricas en manosa-6-fosfato (manosa-6-P), que actúa como un marcador molecular, dirigiendo a las enzimas hacia la ruta de los lisosomas.
La ausencia o disfunción de alguna de las enzimas contenidas en los lisosomas conduce a una amplia variedad de deficiencias enzimáticas que provocan la acumulación de diversas sustancias en el interior de los lisosomas, provocando un amplio abanico de enfermedades cuyos síntomas varían según el tipo de metabolito acumulado.
Actualmente, la terapia de sustitución enzimática (TSE) no ofrece un tratamiento curativo para este tipo de patologías, aunque sí contribuye a paliar la sintomatología asociada, siendo candidatas claras, por su origen monogénico, al desarrollo de terapias génicas específicas.
Esfingolipidosis
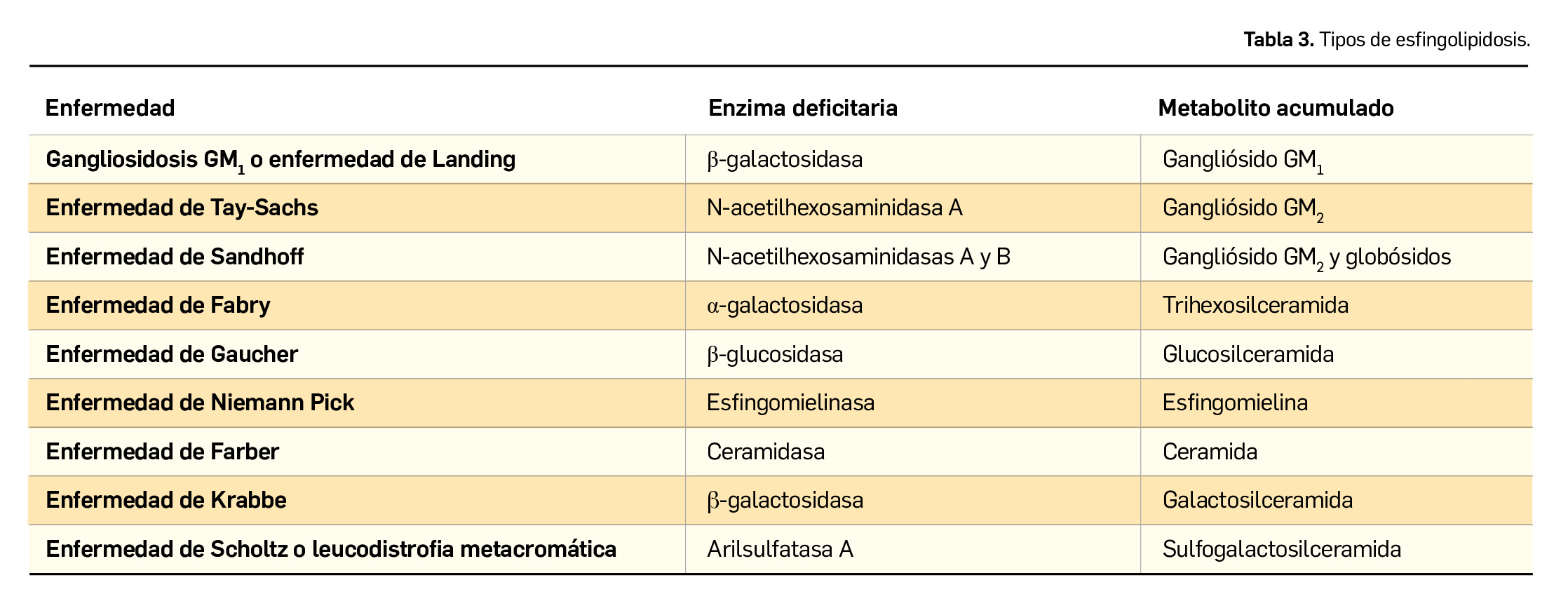
Constituyen un conjunto de enfermedades metabólicas hereditarias producidas por la ausencia total o parcial de las enzimas encargadas de la degradación de esfingolípidos, resultantes de la unión de la ceramida, unidad básica estructural, a un resto glucídico. Las distintas esfingolipidosis se recogen en la Tabla 3.
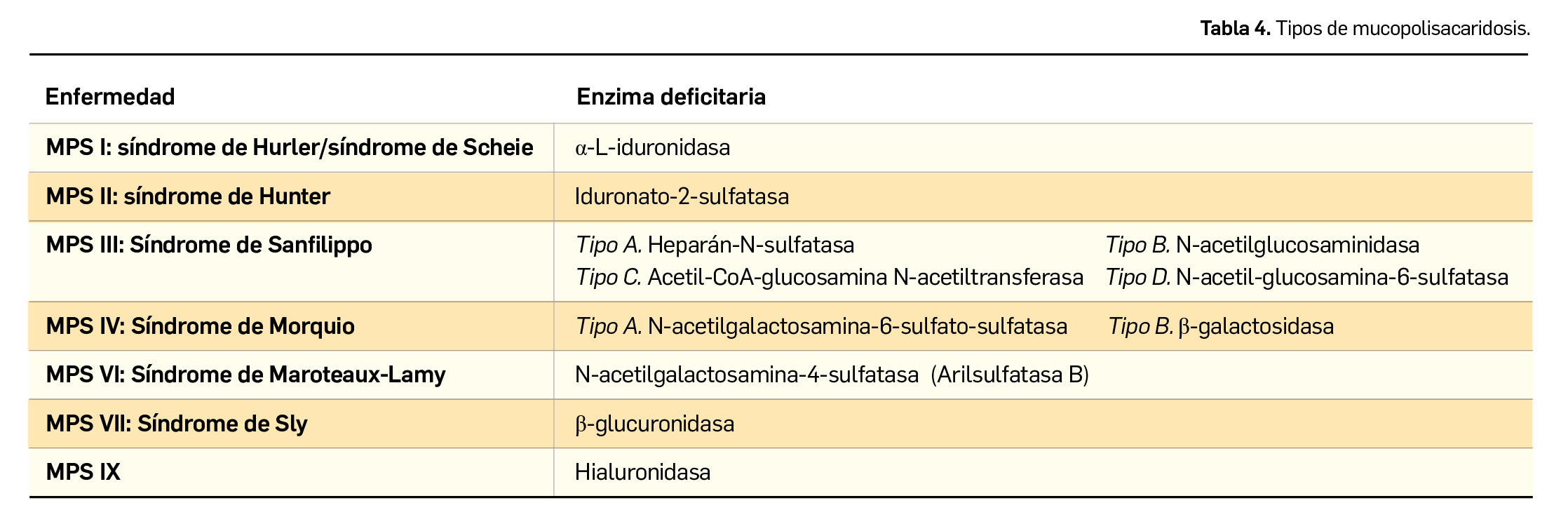
Mucopolisacaridosis
Representan un conjunto de enfermedades producidas por la deficiencia de alguna de las enzimas responsables de la degradación lisosomal de mucopolisacáridos (Tabla 4).
Otras alteraciones de depósito lisosomal
Entre las alteraciones de la degradación lisosomal de glucoproteínas y oligosacáridos pueden citarse la α-manosidosis (déficit de lipasa ácida lisosomal), la fucosidosis (déficit de α fucosidasa) y la sialidosis (déficit de neuraminidasa).
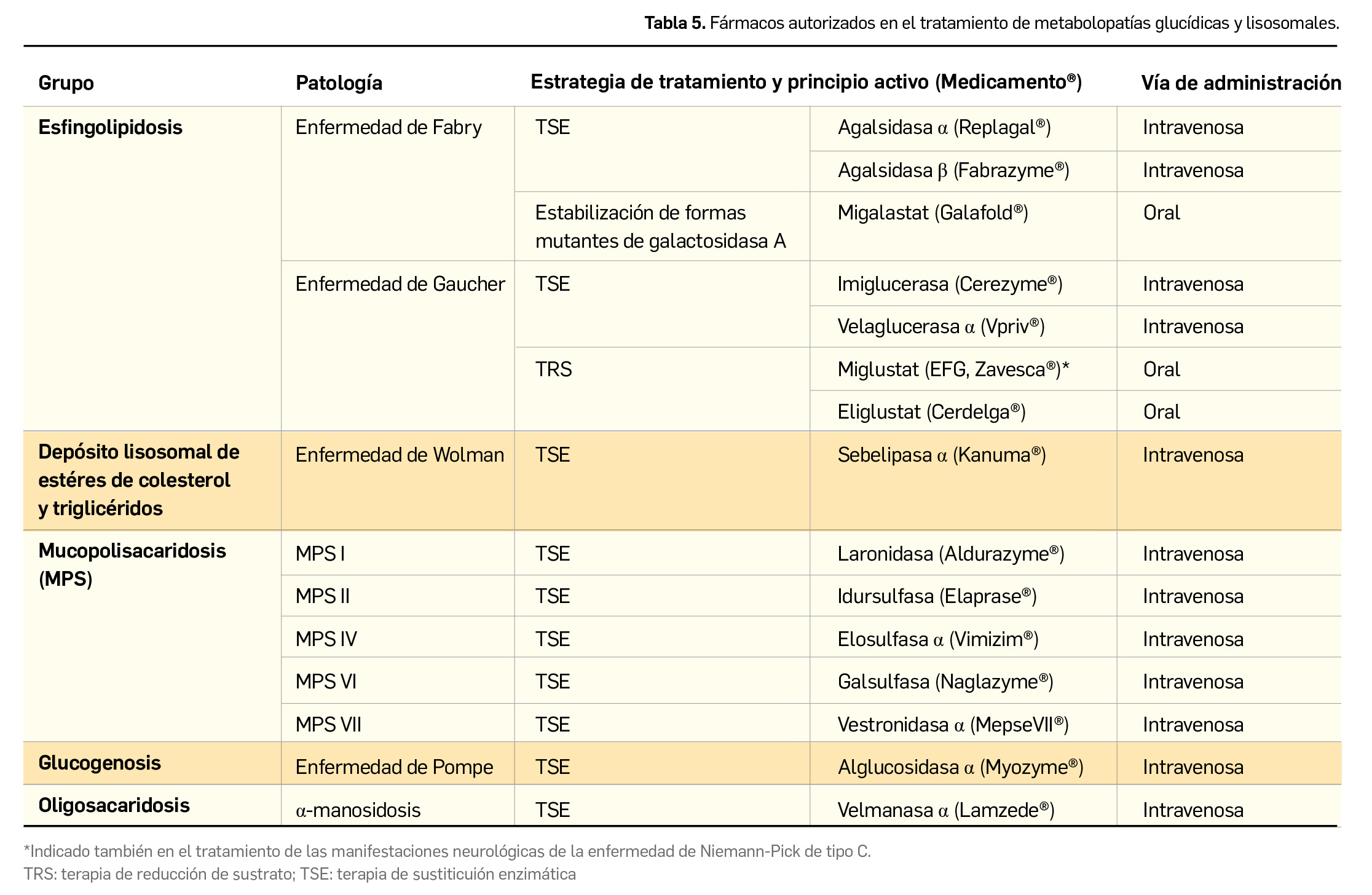
En las metabolopatías de origen lisosomal, el tratamiento (Tabla 5) puede realizarse mediante:
- Terapia de sustitución enzimática (TSE), o administración periódica de una enzima activa a un paciente con un trastorno causado por una deficiencia enzimática, con el objetivo de sustituir la enzima deficitaria, corrigiendo así el defecto metabólico.
- Terapia de reducción de sustrato (TRS), con la consiguiente menor disponibilidad del sustrato biológico sobre el cual actúa la enzima deficitaria, previniendo su acumulación.
Otras metabolopatías
Metabolopatías mitocondriales
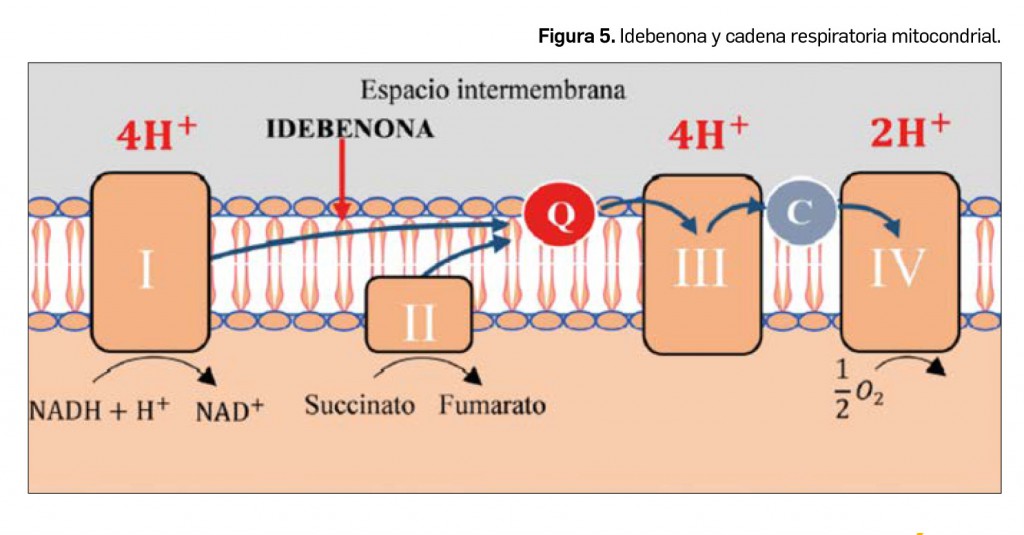
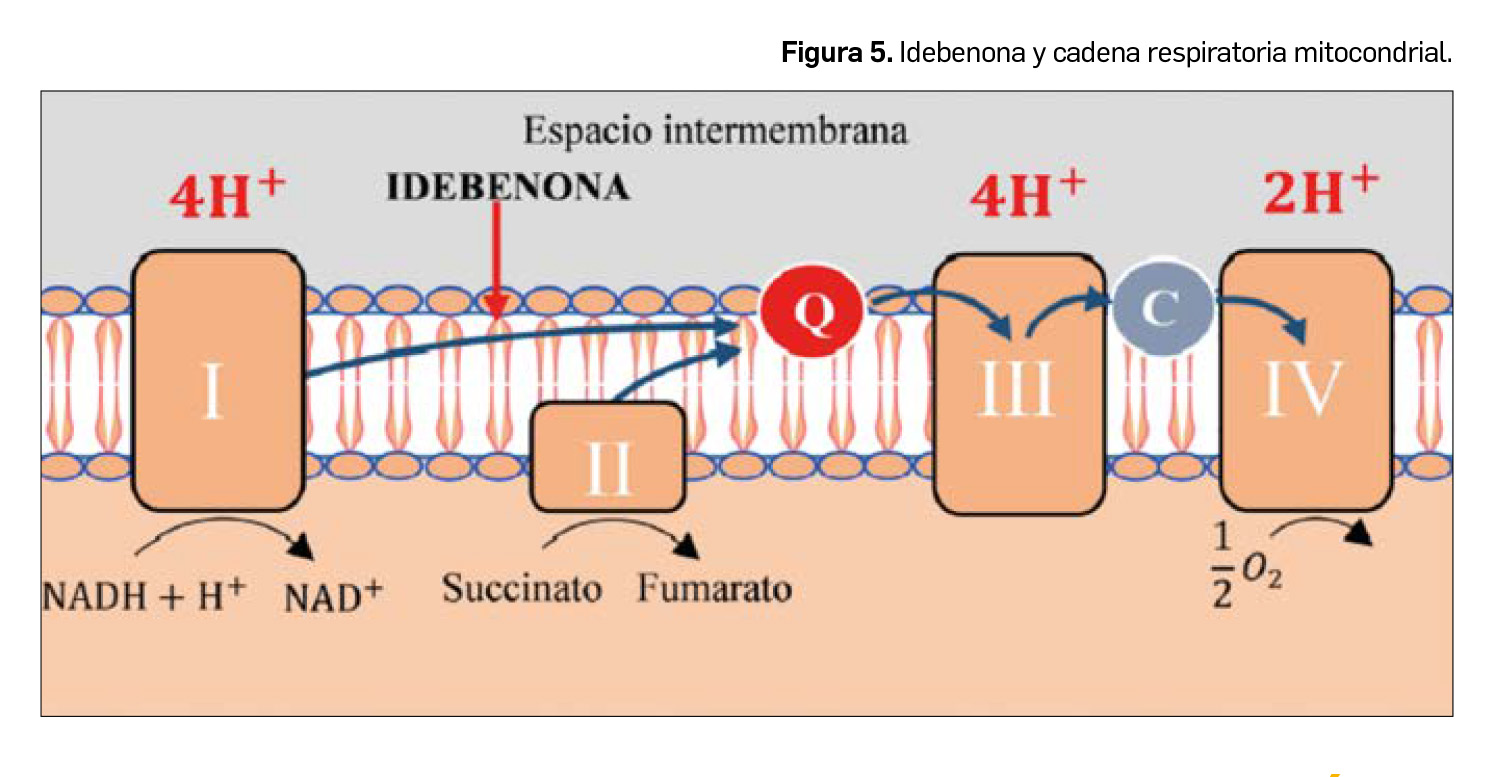
La neuropatía óptica de Leber ocasiona una pérdida súbita de la visión en aquellos pacientes portadores de las mutaciones puntuales ND1, ND4 y ND6 del complejo I de la cadena respiratoria mitocondrial. Se han conseguido buenos resultados con el
tratamiento con idebenona (Figura 5), una benzoquinona análoga de la coenzima Q10 que actúa transfiriendo directamente los electrones desde el complejo I (afectado por las mutaciones) al complejo III de la cadena respiratoria mitocondrial, restaurando así la producción de ATP celular.
Metabolopatías peroxisomales
Se incluyen en este grupo un conjunto de enfermedades hereditarias poco frecuentes, caracterizadas por alteraciones graves en cerebro, riñones, hígado y esqueleto, como el síndrome de Zellweger y la adrenoleucodistrofia ligada al cromosoma X. En este contexto, la mezcla de ácidos grasos de cadena larga insaturados, conocida como aceite de Lorenzo, podría reducir el riesgo de daño cerebral si se administra de manera temprana.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}