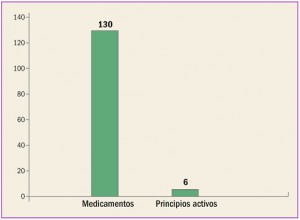
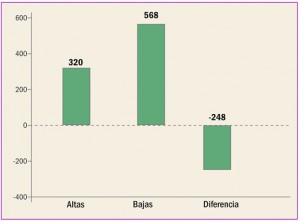
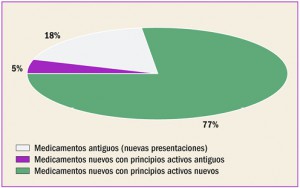
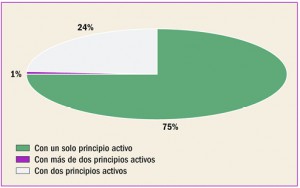
MEDICAMENTOS DADOS DE ALTA (mayo 2017)
|
NP |
CÓDIGO |
NOMBRE, PRESENTACIÓN Y LABORATORIO |
G. TER. |
PRECIO (€) |
DISP. |
TIPO |
|
| 7125112 | ACIDO ACETILSALICILICO TARBIS EFG 100 MG 30 COMPRIMIDOS GASTRORRESISTENTES (TARBIS FARMA, S.L.) | B01AC | 1,45 € | R | |||
| 7121978 | AMLODIPINO/VALSARTAN KERN PHARMA EFG 10/160 MG 28 COMPRIMIDOS RECUBIERTOS (KERN PHARMA) | C09DB | 14,11 € | R | |||
| 7121961 | AMLODIPINO/VALSARTAN KERN PHARMA EFG 5/160 MG 28 COMPRIMIDOS RECUBIERTOS (KERN PHARMA) | C09DB | 13,27 € | R | |||
| 7120872 | AMLODIPINO/VALSARTAN RATIOPHARM EFG 10/160 MG 28 COMPRIMIDOS RECUBIERTOS (RATIOPHARM) | C09DB | 14,11 € | R | |||
| 7120865 | AMLODIPINO/VALSARTAN RATIOPHARM EFG 5/160 MG 28 COMPRIMIDOS RECUBIERTOS (RATIOPHARM) | C09DB | 13,27 € | R | |||
| 7122418 | AMLODIPINO/VALSARTAN TEVA EFG 10/160 MG 28 COMPRIMIDOS RECUBIERTOS (TEVA PHARMA S.L.U.) | C09DB | 14,11 € | R | |||
| 7122395 | AMLODIPINO/VALSARTAN TEVA EFG 5/160 MG 28 COMPRIMIDOS RECUBIERTOS (TEVA PHARMA S.L.U.) | C09DB | 13,27 € | R | |||
| N | 7142874 | BELKYRA 10 MG/ML 4 VIALES INYECTABLE 2 ML (ALLERGAN S. A.) | D11AX | 952,55 € | R | H | |
| 7123484 | BETADINE EUROMEDICINES 100 MG/ML SOLUCION TOPICA 1 FRASCO 125 ML (EUROMEDICINES) | D08AG | 5,53 € | MSP | |||
| 7136248 | BISOPROLOL PENSA EFG 10 MG 28 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 3,06 € | R | AR | ||
| 7136255 | BISOPROLOL PENSA EFG 10 MG 30 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 3,28 € | R | AR | ||
| 7136262 | BISOPROLOL PENSA EFG 10 MG 60 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 6,56 € | R | AR | ||
| 7136163 | BISOPROLOL PENSA EFG 2.5 MG 28 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 2,40 € | R | AR | ||
| 7136187 | BISOPROLOL PENSA EFG 5 MG 28 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 2,50 € | R | AR | ||
| 7136194 | BISOPROLOL PENSA EFG 5 MG 30 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 2,36 € | R | AR | ||
| 7136200 | BISOPROLOL PENSA EFG 5 MG 60 COMPRIMIDOS (BLISTER PVC/PVDC) (PENSA PHARMA S.A.) | C07AB | 3,28 € | R | AR | ||
| 7017394 | BLUXAM 20 MG/ML 10 AMPOLLAS SUSPENSION INYECTABLE 1 ML (ROVI) | H02AB | 40,59 € | R | AR | ||
| 7112518 | BRALTUS 10 MCG 30 CAPSULAS + 1 INHALADOR ZONDA (TEVA PHARMA S.L.U.) | R03BB | 39,25 € | R | AR | ||
| 7146308 | CASPOFUNGINA TEVAGEN EFG 50 MG 1 VIAL CONCENTRADO PERFUSION 10 ML (TEVA PHARMA S.L.U.) | J02AX | 332,98 € | FR R | H | ||
| 7146292 | CASPOFUNGINA TEVAGEN EFG 70 MG 1 VIAL CONCENTRADO PERFUSION 10 ML (TEVA PHARMA S.L.U.) | J02AX | 409,14 € | FR R | H | ||
| 7104131 | CINACALCET ACCORD EFG 30 MG 28 COMPRIMIDOS RECUBIERTOS (ACCORD HEALTHCARE, S.L.U.) | H05BX | 159,50 € | R | H | ||
| 7104148 | CINACALCET ACCORD EFG 60 MG 28 COMPRIMIDOS RECUBIERTOS (ACCORD HEALTHCARE, S.L.U.) | H05BX | 253,93 € | R | H | ||
| 7104162 | CINACALCET ACCORD EFG 90 MG 28 COMPRIMIDOS RECUBIERTOS (ACCORD HEALTHCARE, S.L.U.) | H05BX | 362,21 € | R | H | ||
| 7135555 | DEXAMETASONA KRKA EFG 4 MG 30 COMPRIMIDOS (KRKA FARMACEUTICA S.L.) | H02AB | 11,99 € | R | |||
| 7135562 | DEXAMETASONA KRKA EFG 8 MG 30 COMPRIMIDOS (KRKA FARMACEUTICA S.L.) | H02AB | 19,19 € | R | |||
| 6936191 | DONEPEZILO SANOVEL EFG 10 MG 28 COMPRIMIDOS RECUBIERTOS (SANOVEL SPAIN S.L.) | N06DA | 65,16 € | R | DH CPD | ||
| NP | 7068242 | EPORATIO 10000 UI 6 JERINGAS PRECARGADAS CON AGUJA 1 ML (TEVA PHARMA S.L.U.) | B03XA | 417,67 € | FR R | H | |
| 7032250 | EPROSARTAN MYLAN PHARMACEUTICALS EFG 600 MG 28 COMPRIMIDOS RECUBIERTOS (MYLAN PHARMACEUTICALS S.L.) | C09CA | 14,27 € | R | AR | ||
| NP | 7001324 | ESCITALOPRAM ALTER GENERICOS EFG 20 MG 56 COMPRIMIDOS RECUBIERTOS (ALTER) | N06AB | 35,08 € | R | AR | |
| NP | 7112471 | EXJADE 360 MG 30 COMPRIMIDOS RECUBIERTOS (NOVARTIS FARMACEUTICA) | V03AC | 746,11 € | R | DH CPD AR | |
| NP | 7112488 | EXJADE 90 MG 30 COMPRIMIDOS RECUBIERTOS (NOVARTIS FARMACEUTICA) | V03AC | 219,74 € | R | DH CPD AR | |
| NP | 6942918 | FINASTERIDA CINFA EFG 1 MG 1 MG 98 COMPRIMIDOS RECUBIERTOS (CINFA) | D11AX | 58,90 € | R | EXO | |
| 7073420 | GARDASIL 9 1 JERINGA PRECARGADA 0.5 ML (MSD) | J07BM | 172,55 € | FR R | DIHSC | ||
| 7120841 | GLICLAZIDA RATIOPHARM EFG 60 MG 60 COMP LIB MODIFICADA (PVC/AL) (RATIOPHARM) | A10BB | 11,18 € | R | AR | ||
| NP | 7120810 | GLICLAZIDA TEVA EFG 60 MG 60 COMP LIB MODIFICADA (PVC/AL) (TEVA PHARMA S.L.U.) | A10BB | 11,18 € | R | AR | |
| 7141259 | GRANPIDAM EFG 20 MG 90 COMPRIMIDOS RECUBIERTOS (ACCORD HEALTHCARE, S.L.U.) | G04BE | 359,76 € | R | H | ||
| NP | 6967836 | IRBESARTAN/HIDROCLOROTIAZIDA MYLAN EFG 300/12.5 MG 28 COMPRIMIDOS RECUBIERTOS (MYLAN PHARMACEUTICALS S.L.) | C09DA | 19,29 € | R | ||
| 7141747 | IVABRADINA NORMON EFG 5 MG 56 COMPRIMIDOS RECUBIERTOS (NORMON) | C01EB | 42,24 € | R | AR | ||
| 7141754 | IVABRADINA NORMON EFG 7.5 MG 56 COMPRIMIDOS RECUBIERTOS (NORMON) | C01EB | 43,27 € | R | AR | ||
| 7140726 | IVABRADINA RATIO EFG 5 MG 56 COMPRIMIDOS RECUBIERTOS (RATIOPHARM) | C01EB | 42,24 € | R | AR | ||
| 7140733 | IVABRADINA RATIO EFG 7.5 MG 56 COMPRIMIDOS RECUBIERTOS (RATIOPHARM) | C01EB | 43,27 € | R | AR | ||
| 7142591 | IVABRADINA SANDOZ EFG 5 MG 56 COMPRIMIDOS RECUBIERTOS (SANDOZ FARMACEUTICA S.A.) | C01EB | 42,24 € | R | AR | ||
| 7142607 | IVABRADINA SANDOZ EFG 7.5 MG 56 COMPRIMIDOS RECUBIERTOS (SANDOZ FARMACEUTICA S.A.) | C01EB | 43,27 € | R | AR | ||
| NP | 7148968 | LEVETIRACETAM NORMON EFG 1000 MG 60 COMPRIMIDOS RECUBIERTOS (NORMON) | N03AX | 109,09 € | R | AR | |
| NP | 7103059 | LEVOFLOXACINO PHARMA COMBIX EFG 500 MG 7 COMPRIMIDOS RECUBIERTOS (COMBIX S.L.) | J01MA | 13,11 € | R | ||
| NP | 7106258 | MOVICOL “13.8 G” 10 SOBRES SOLUCION ORAL 25 ML (NORGINE DE ESPAÑA) | A06AD | 8,43 € | R | EXO | |
| NP | 7106272 | MOVICOL “13.8 G” 30 SOBRES SOLUCION ORAL 25 ML (NORGINE DE ESPAÑA) | A06AD | 16,20 € | R | EXO | |
| 7137108 | NORDIMET 15 MG/0.6 ML 1 PLUMA PRECARGADA 0.6 ML (NORDIC PHARMA S.A.U.) | L01BA | 21,98 € | R | AR | ||
| 7137115 | NORDIMET 17.5 MG/0.7 ML 1 PLUMA PRECARGADA 0.7 ML (NORDIC PHARMA S.A.U.) | L01BA | 25,96 € | R | AR | ||
| 7137122 | NORDIMET 20 MG/0.8 ML 1 PLUMA PRECARGADA 0.8 ML (NORDIC PHARMA S.A.U.) | L01BA | 26,62 € | R | AR | ||
| 7137139 | NORDIMET 22.5 MG/0.9 ML 1 PLUMA PRECARGADA 0.9 ML (NORDIC PHARMA S.A.U.) | L01BA | 30,60 € | R | AR | ||
| 7137146 | NORDIMET 25 MG/1 ML 1 PLUMA PRECARGADA 1 ML (NORDIC PHARMA S.A.U.) | L01BA | 28,44 € | R | AR | ||
| 6526477 | NOVIDOL 50 MG/G GEL TOPICO 1 TUBO 60 G (MENARINI) | M02AA | 8,93 € | EXO MSP | |||
| NP | 7128861 | OMEPRAZOL STADA 40 MG 56 CAPSULAS GASTRORRESISTENTES (FRASCO) (STADA S.L.) | A02BC | 8,30 € | R | ||
| NP | 7129097 | OMEPRAZOL STADA EFG 20 MG 56 CAPSULAS GASTRORRESISTENTES (FRASCO) (STADA S.L.) | A02BC | 4,15 € | R | ||
| 6944394 | PIOGLITAZONA ACCORD EFG 30 MG 28 COMPRIMIDOS (ACCORD HEALTHCARE, S.L.U.) | A10BG | 30,07 € | R | CPD AR | ||
| 7110545 | RABIPUR 1 VIAL POLVO + 1 JER PREC DISOL + 2 AGUJAS (GLAXO SMITHKLINE) | J07BG | 77,63 € | FR R | H | ||
| NP | 7071723 | SILDENAFILO CINFA EFG 50 MG 8 COMPRIMIDOS RECUBIERTOS (CINFA) | G04BE | 46,52 € | R | EXO | |
| N | 7112570 | UPTRAVI 1000 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| N | 7112587 | UPTRAVI 1200 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| N | 7112600 | UPTRAVI 1400 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| N | 7112662 | UPTRAVI 1600 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| N | 7112679 | UPTRAVI 400 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| N | 7112686 | UPTRAVI 600 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| N | 7112693 | UPTRAVI 800 MCG 60 COMPRIMIDOS RECUBIERTOS (ACTELION PHARMACEUTICALS ESPAÑA) | B01AC | 4076,71 € | R | H | |
| 6066980 | ZALVISO 15 MCG 40 COMP SUBLINGUALES (20 CARTUCHOS) (GRÜNENTHAL PHARMA) | N01AH | 2724,68 € | R | E H EXO EC | ||
| N | 7021377 | ZEVTERA 500 MG 10 VIALES POLVO (BASILEA MEDICAL LTD) | J01DI | 615,59 € | FR R | ||
| NP | 7139713 | ZYTIGA 500 MG 60 COMPRIMIDOS (JANSSEN-CILAG) | L02BX | 3594,15 € | R | DH DIHSC |
SIGLAS EMPLEADAS
A: Psicótropo (Anexo II del R.D. 2829/1977 de 6 de octubre).
AR: Aportación Reducida.
CPD75: Visado de inspección >75 años.
CPD: Cupón precinto diferenciado.
DH: Medicamento de Diagnóstico Hospitalario.
DiHSC: Dispensación hospitalaria sin cupón precinto.
E: Estupefaciente.
ECM: Medicamento Control Médico.
EFG: Medicamento Farmacéutico Genérico.
EXO: Excluida Oferta Seguridad Social.
EXOI: Excluida, con cupón precinto diferenciado.
FR: Medicamentos que precisan conservación en frigorífico.
H: Medicamento Hospitalario.
MSP: Medicamento susceptible publicidad al público.
N: Medicamentos con principio activo nuevo.
NP: Nueva presentación.
P: Psicótropo (Anexo I del R.D. 2829/1977 de 6 de octubre).
R: Receta.
ST: Suspensión temporal de comercialización.
TLD: Medicamento de dispensación renovable.

{kind=link}
{kind=link}
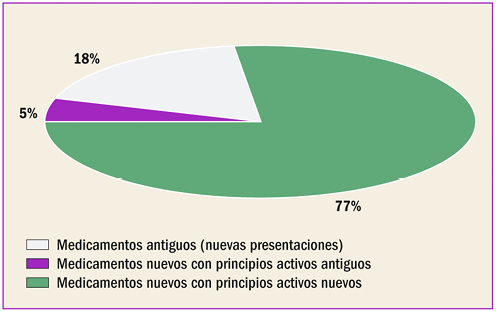{kind=link}
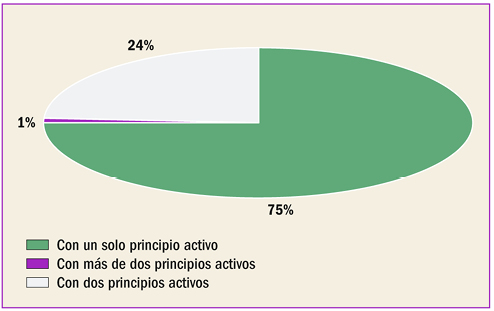{kind=link}
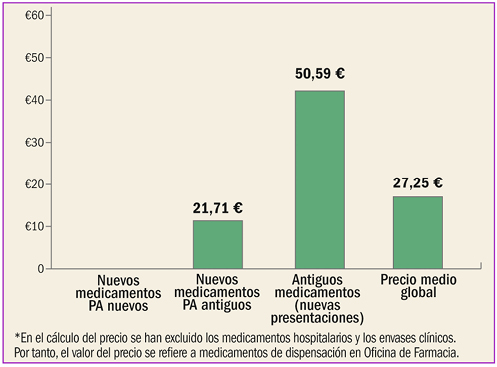{kind=link}
{kind=link}
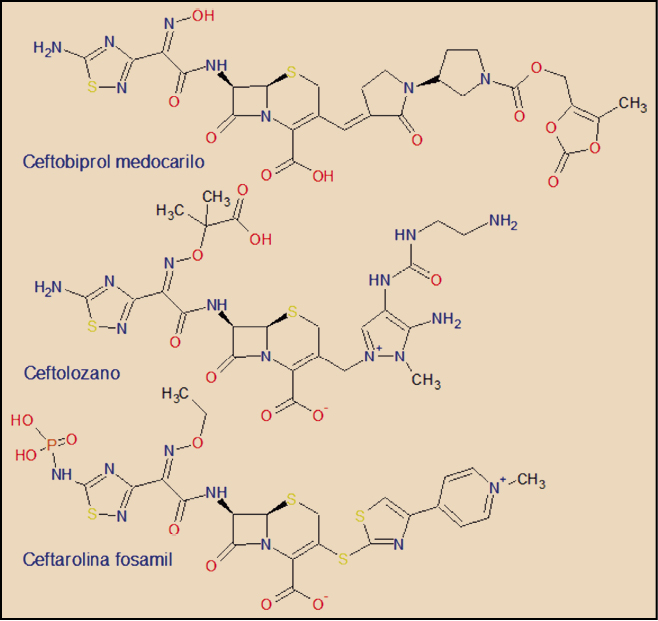{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}