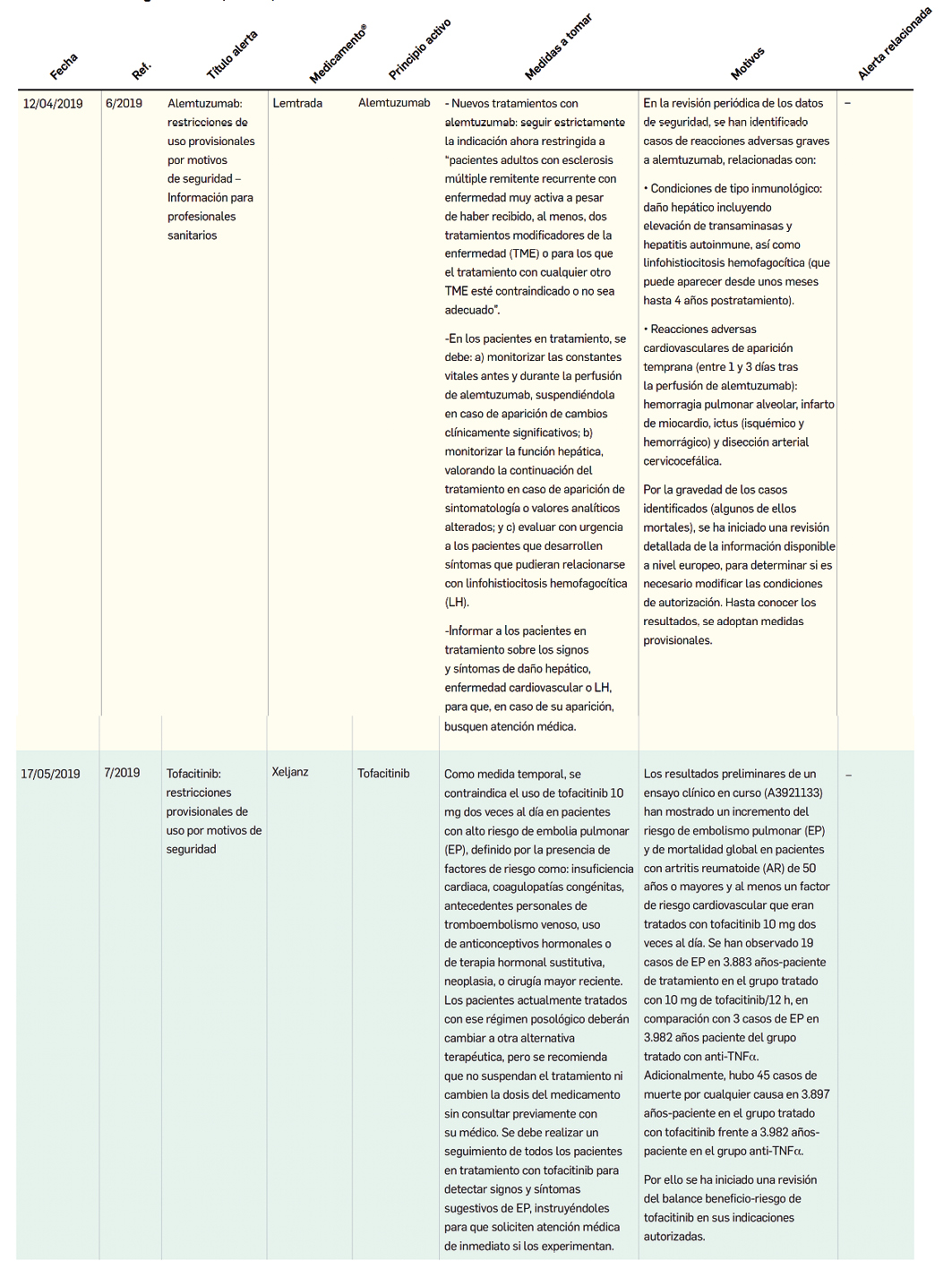
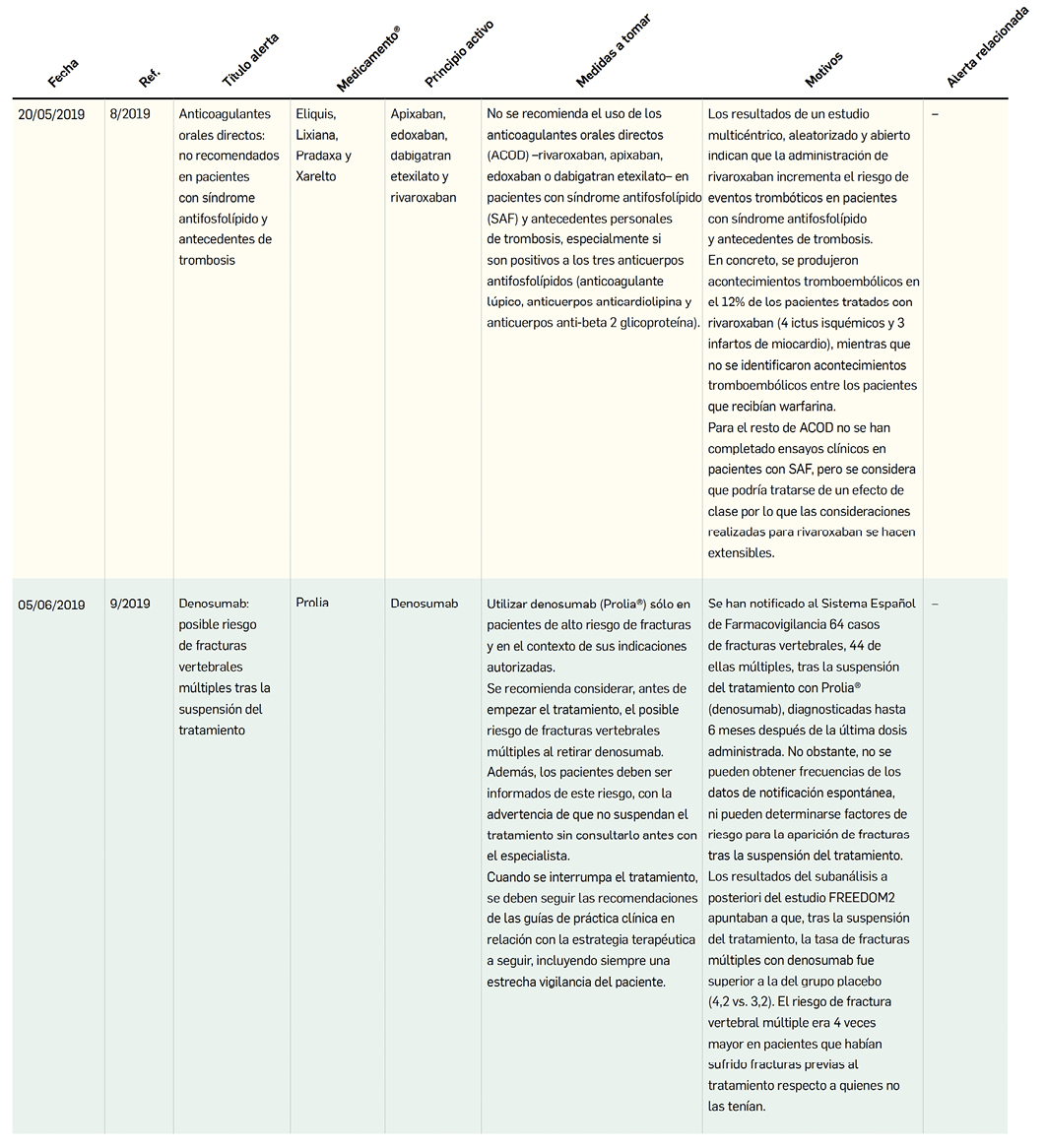
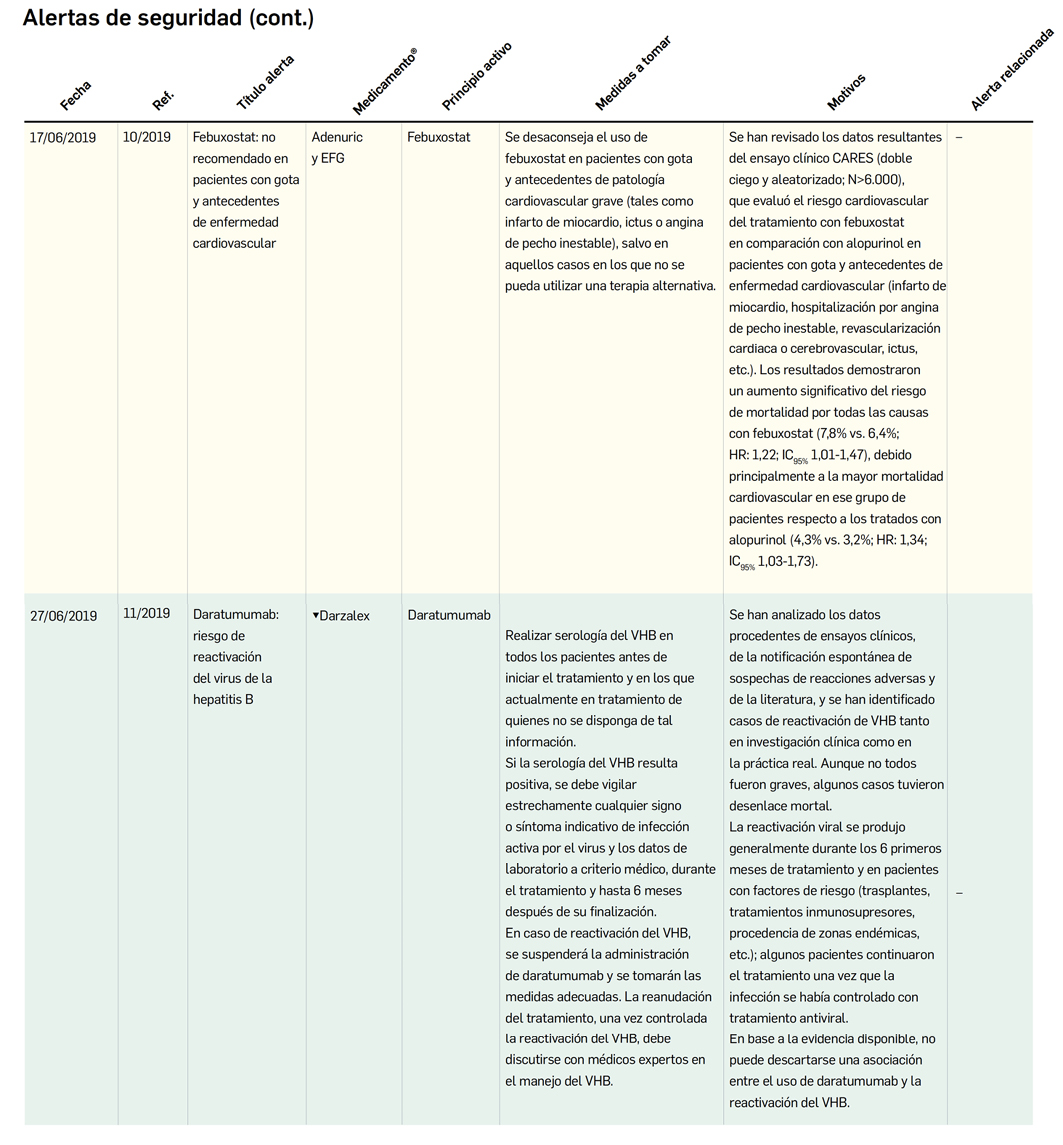
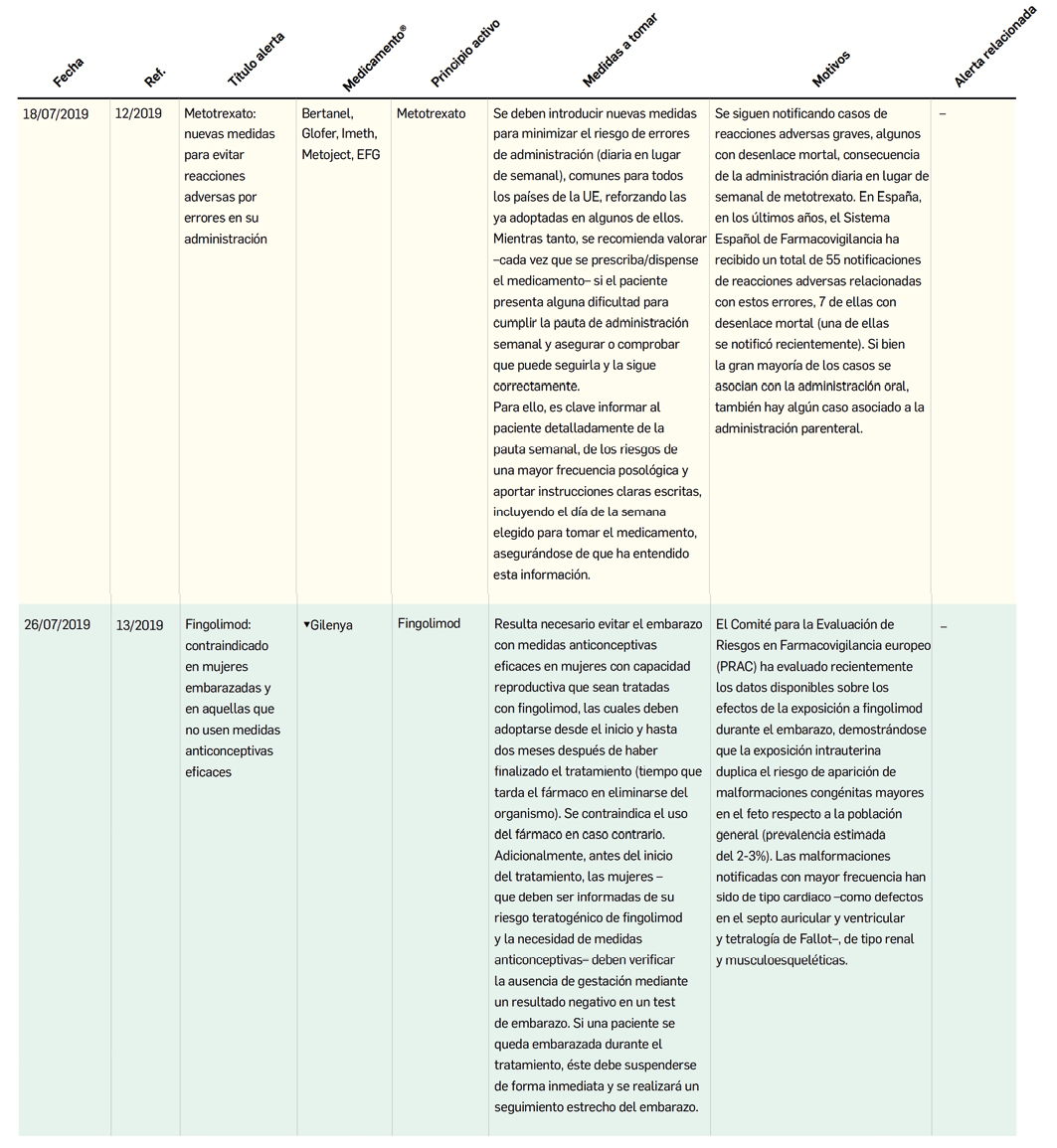
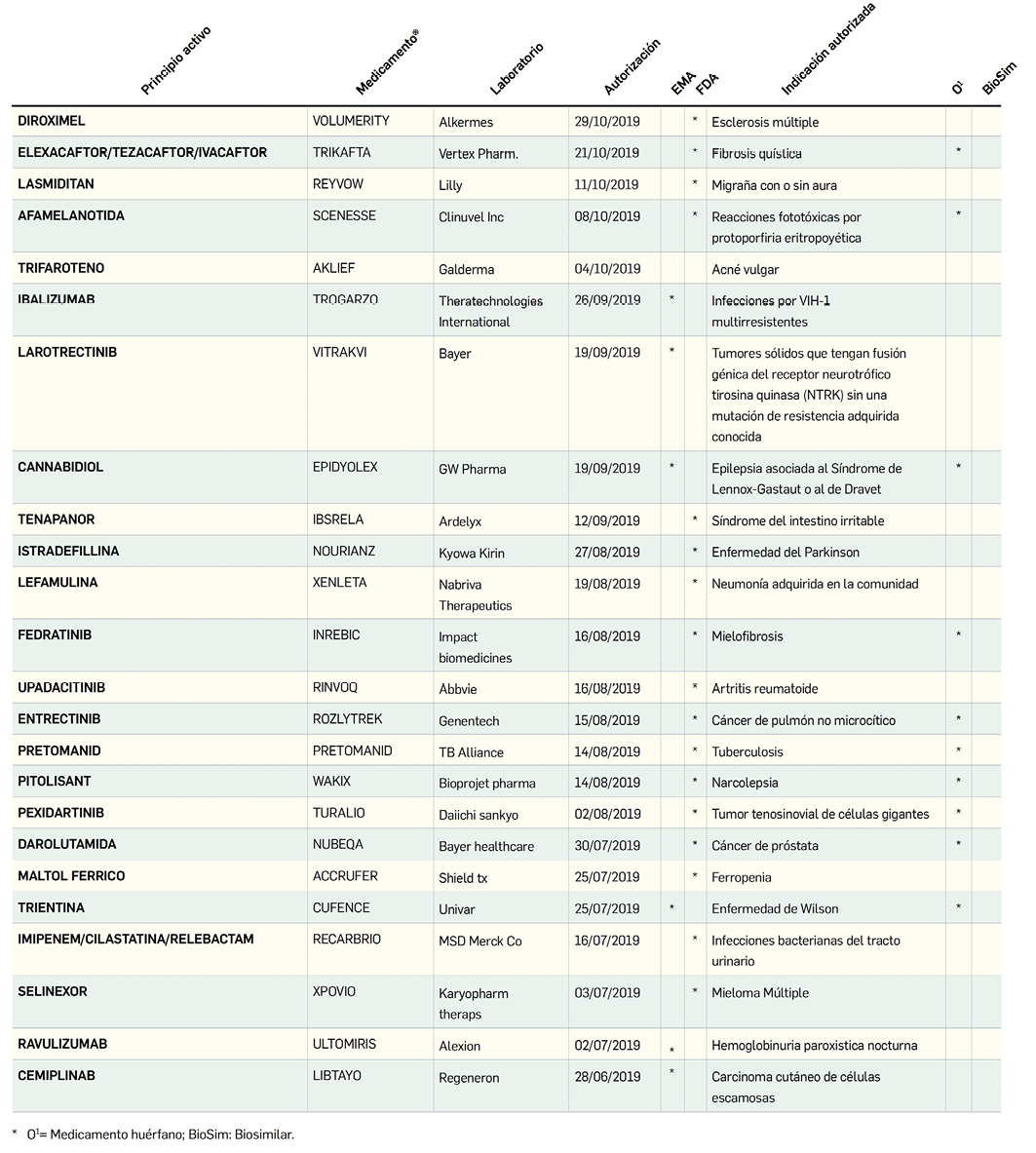
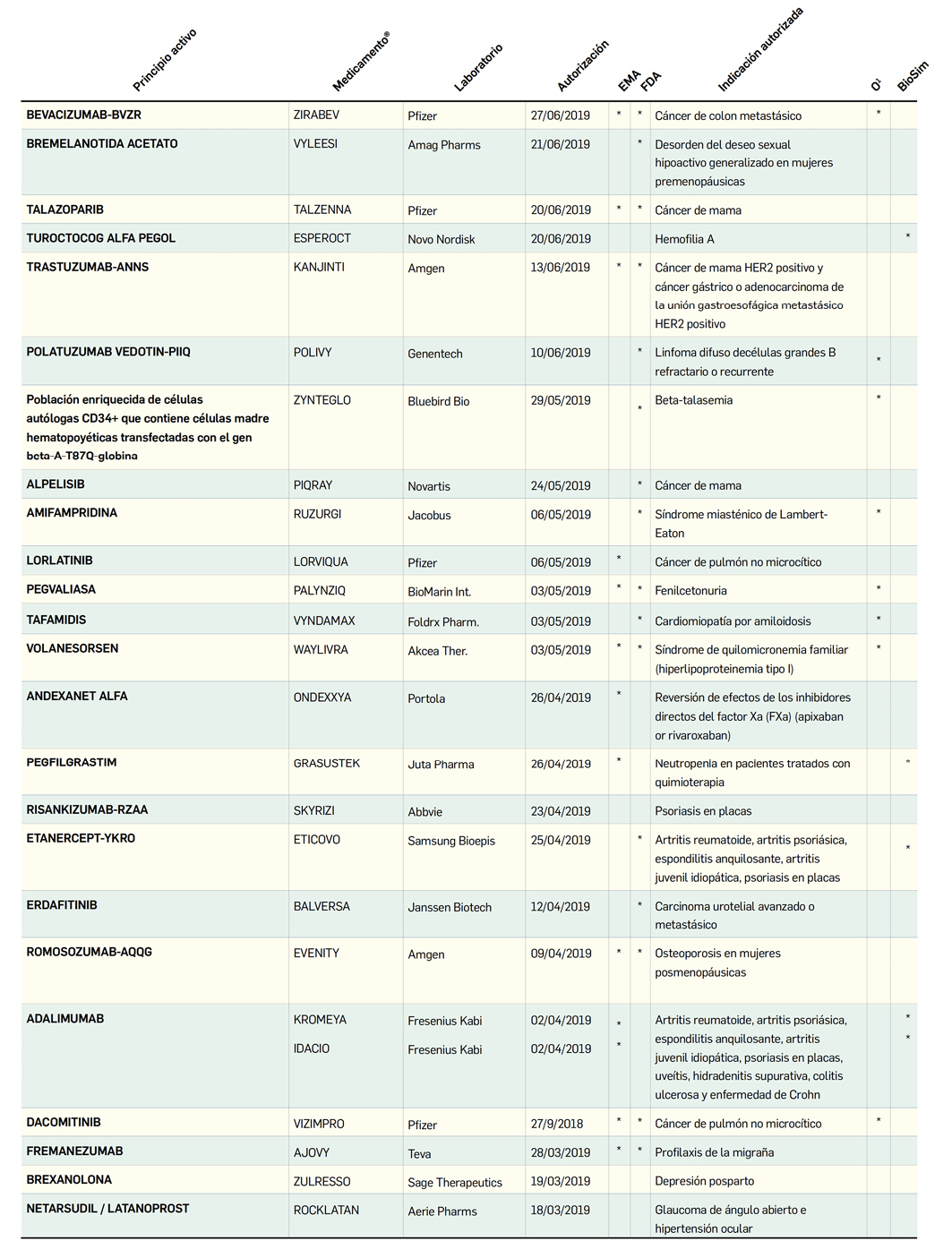
MEDICAMENTOS DE USO HUMANO AUTORIZADOS EN LA UNIÓN EUROPEA (EMA) Y ESTADOS UNIDOS (FDA) DURANTE LOS ÚLTIMOS DOCE MESES, CON NUEVOS PRINCIPIOS ACTIVOS
o biosimilares QUE AÚN NO ESTÁN COMERCIALIZADOS EN ESPAÑA
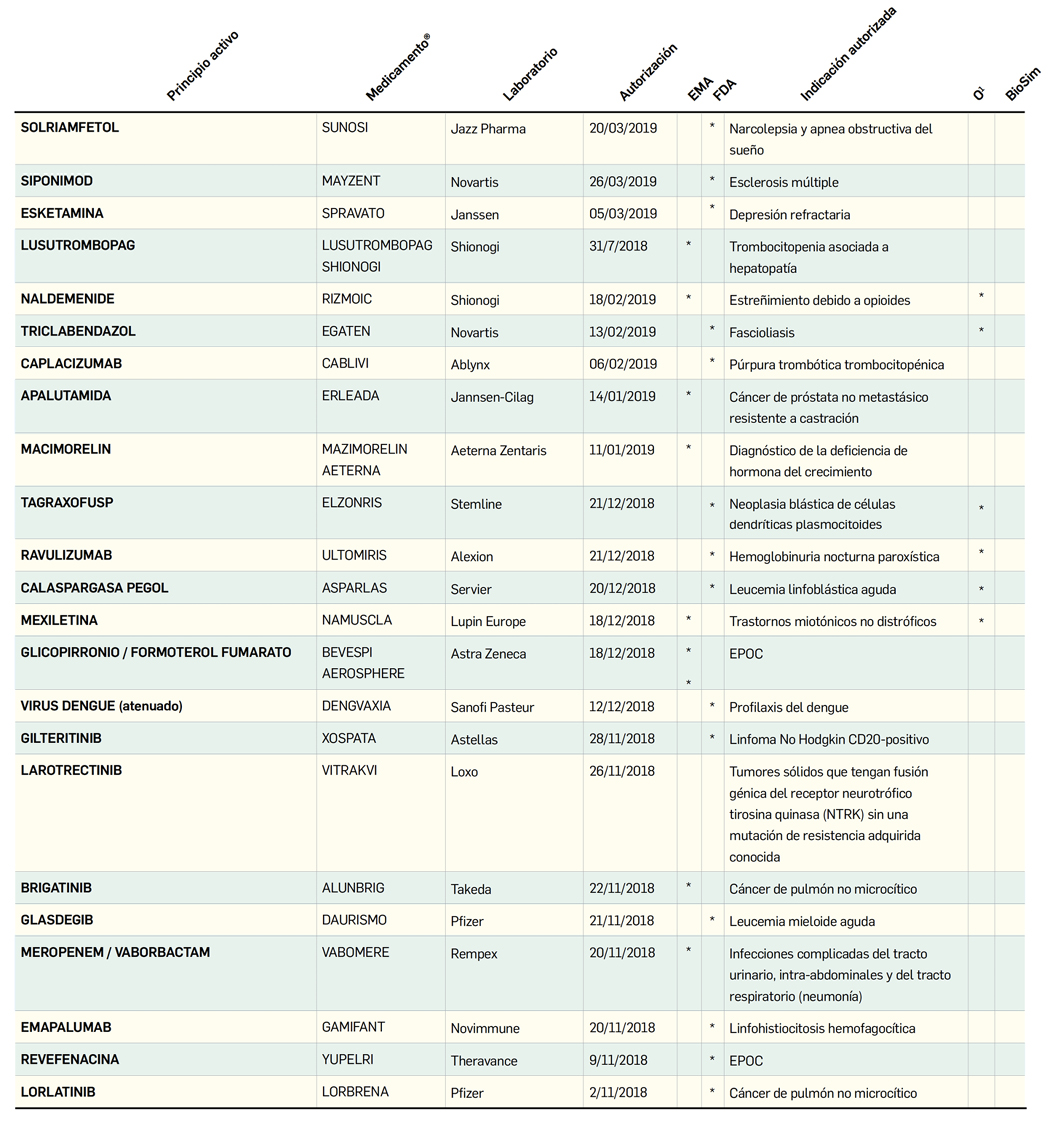
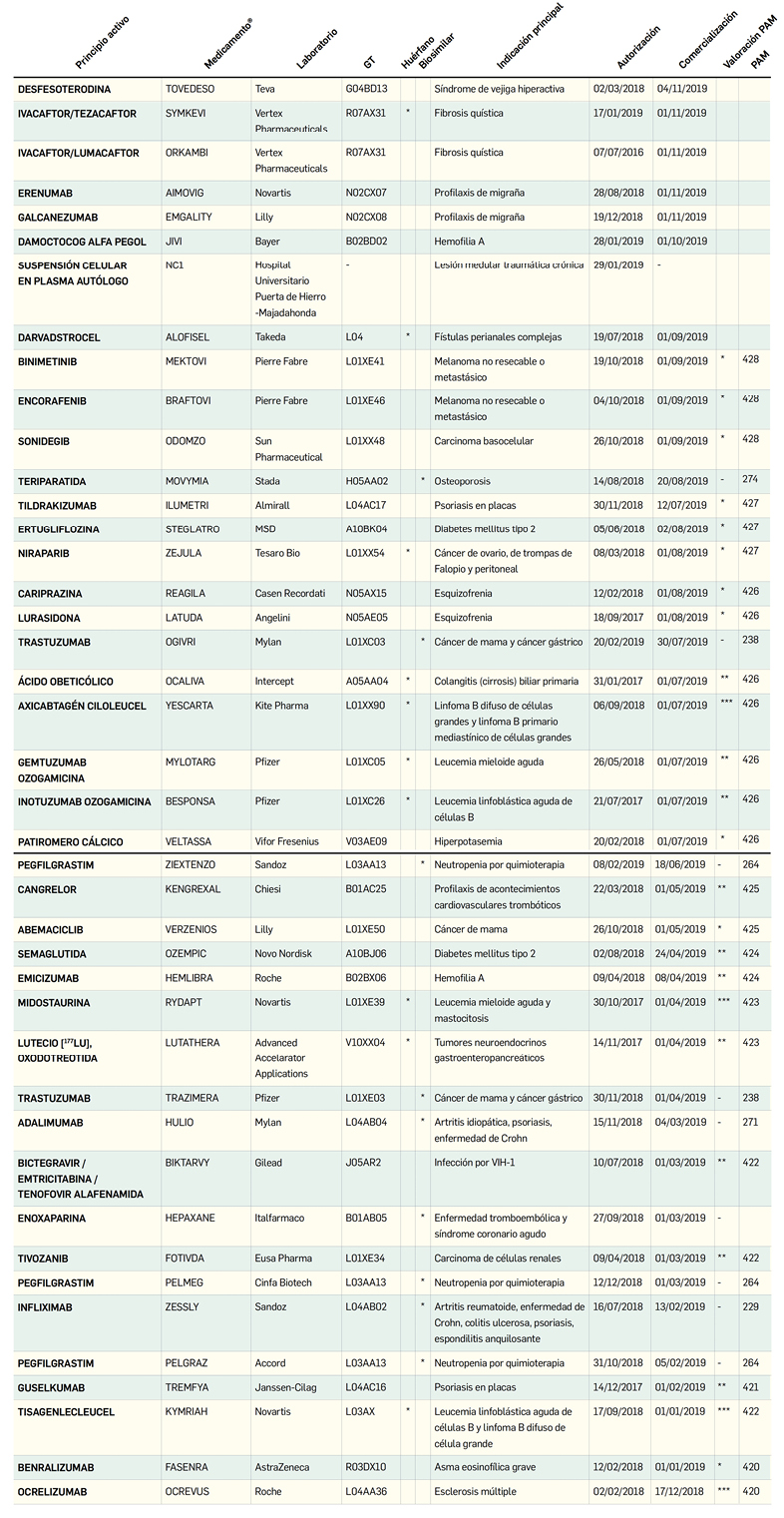
Número 428, Noviembre 2019
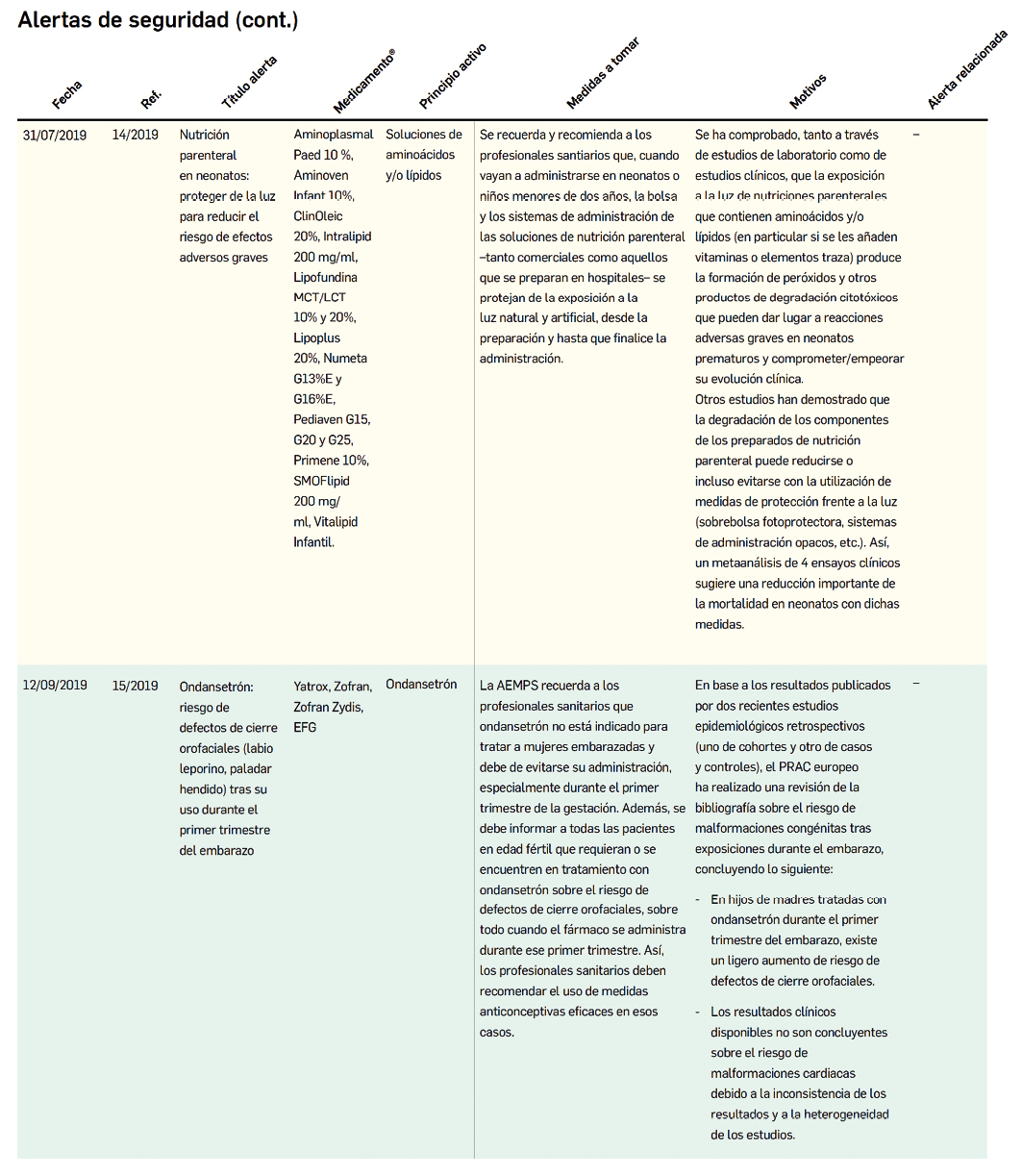
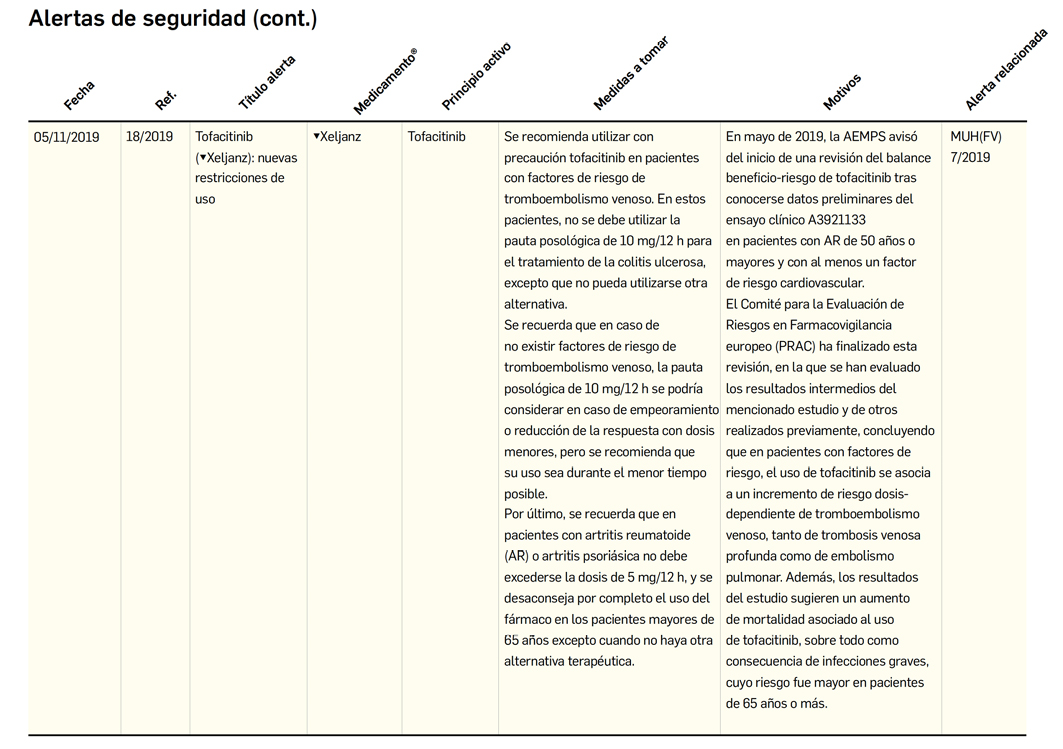
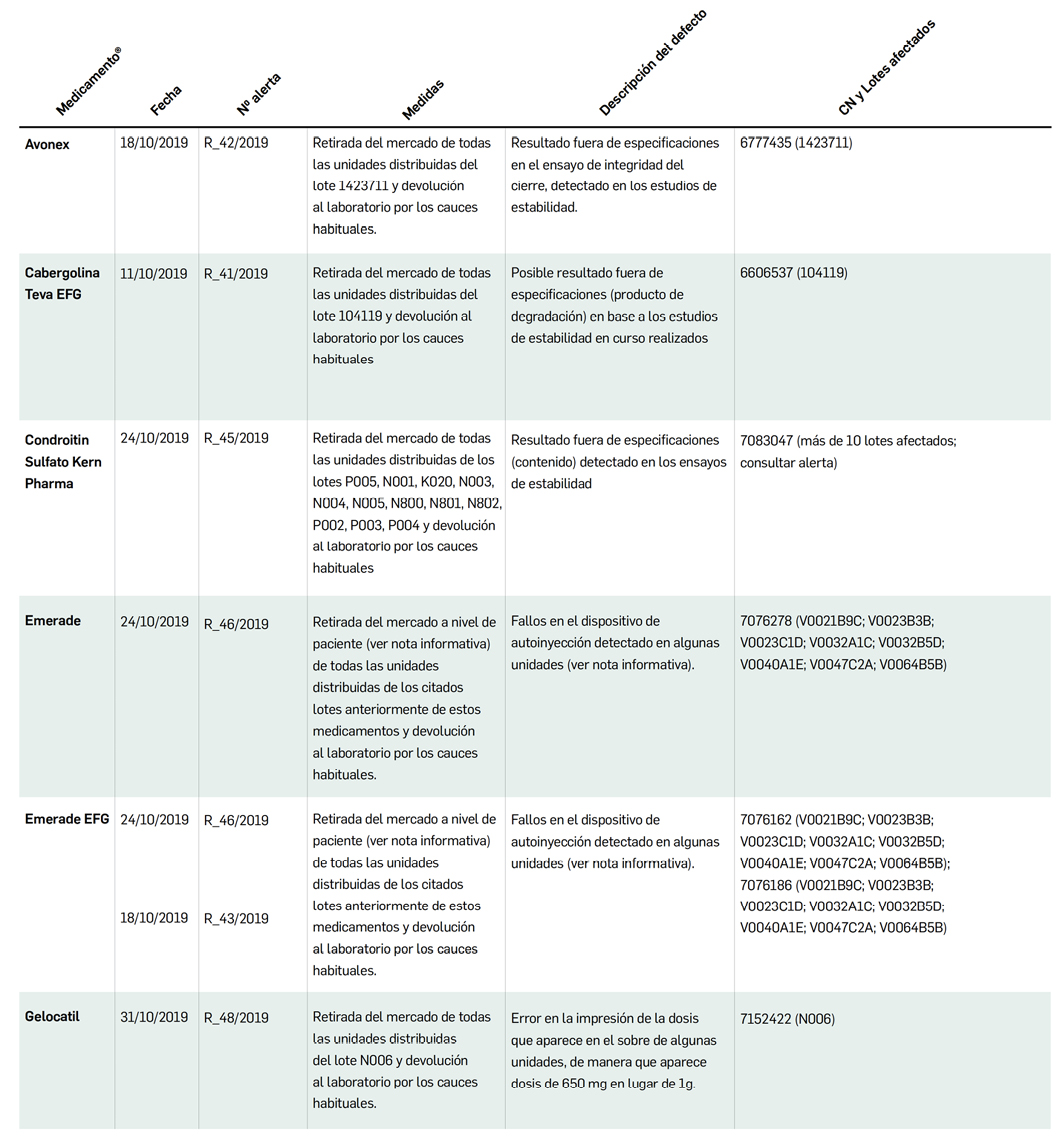
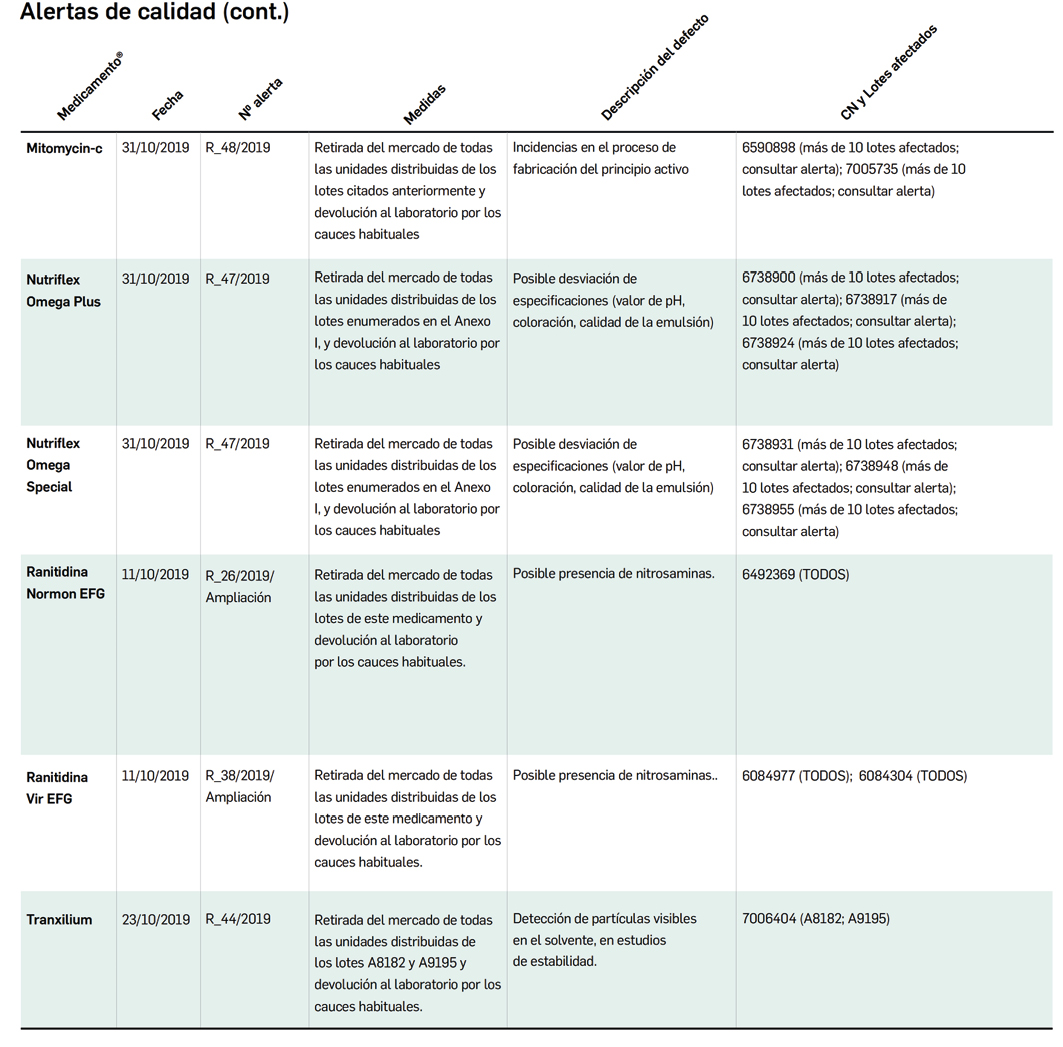
Alertas debidas a defectos de calidad observados en medicamentos de uso humano, publicadas por la AEMPS desde el anterior número y que suponen la retirada o inmovilización de ciertos lotes de medicamentos. En Bot PLUS puede encontrar más información detallada, con acceso al documento de la AEMPS

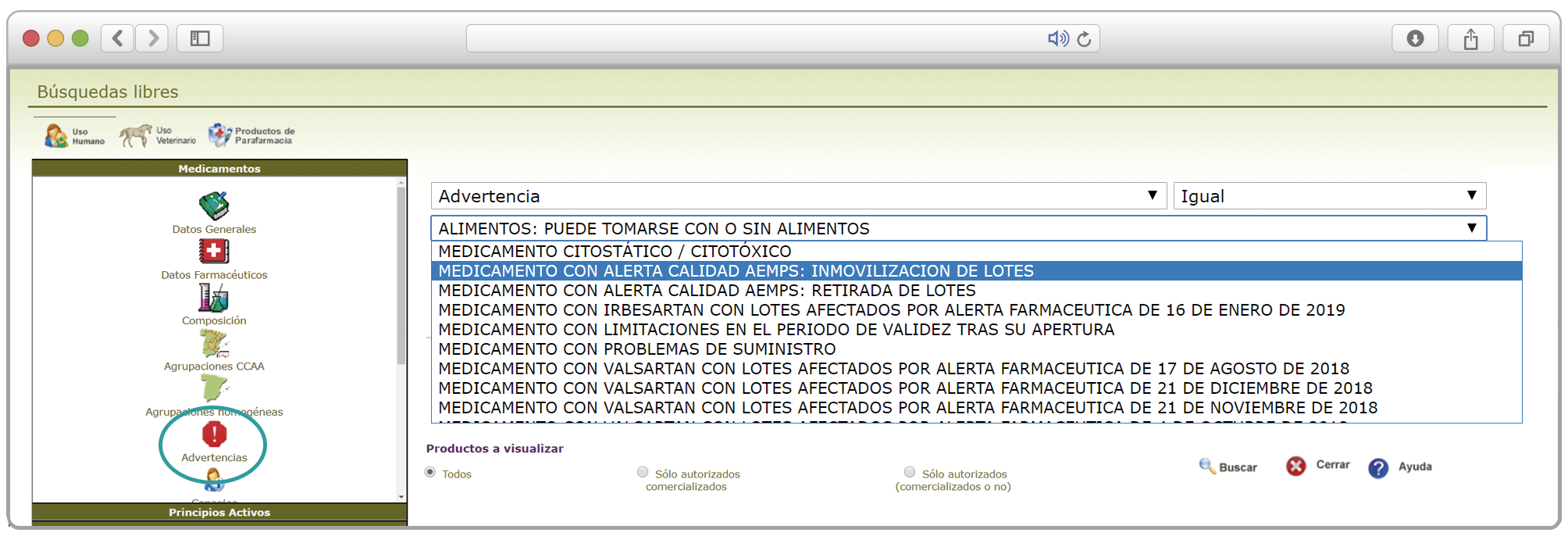
Además de los listados mensuales que podemos consultar en PAM, en Bot PLUS se incorpora la información que publica la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) relativa a notificaciones sobre seguridad y/o calidad de los medicamentos. Mediante un pictograma específico se puede visualizar de forma rápida medicamentos afectados por alguna alerta de seguridad o de defectos de calidad, con tan solo entrar en su ficha.
Al acceder a la ficha de un medicamento afectado por una retirada, se visualiza un mensaje con la advertencia “Medicamento con lotes retirados por Alerta AEMPS.
Ver más información en “Alertas Calidad”.

Además, se incluye una pestaña específica en la que se puede consultar los lotes concretos que han sido retirados, con sus respectivas fechas de caducidad, así como la descripción del defecto de calidad detectado y las medidas a adoptar. También se cuenta con acceso al documento publicado por la AEMPS.
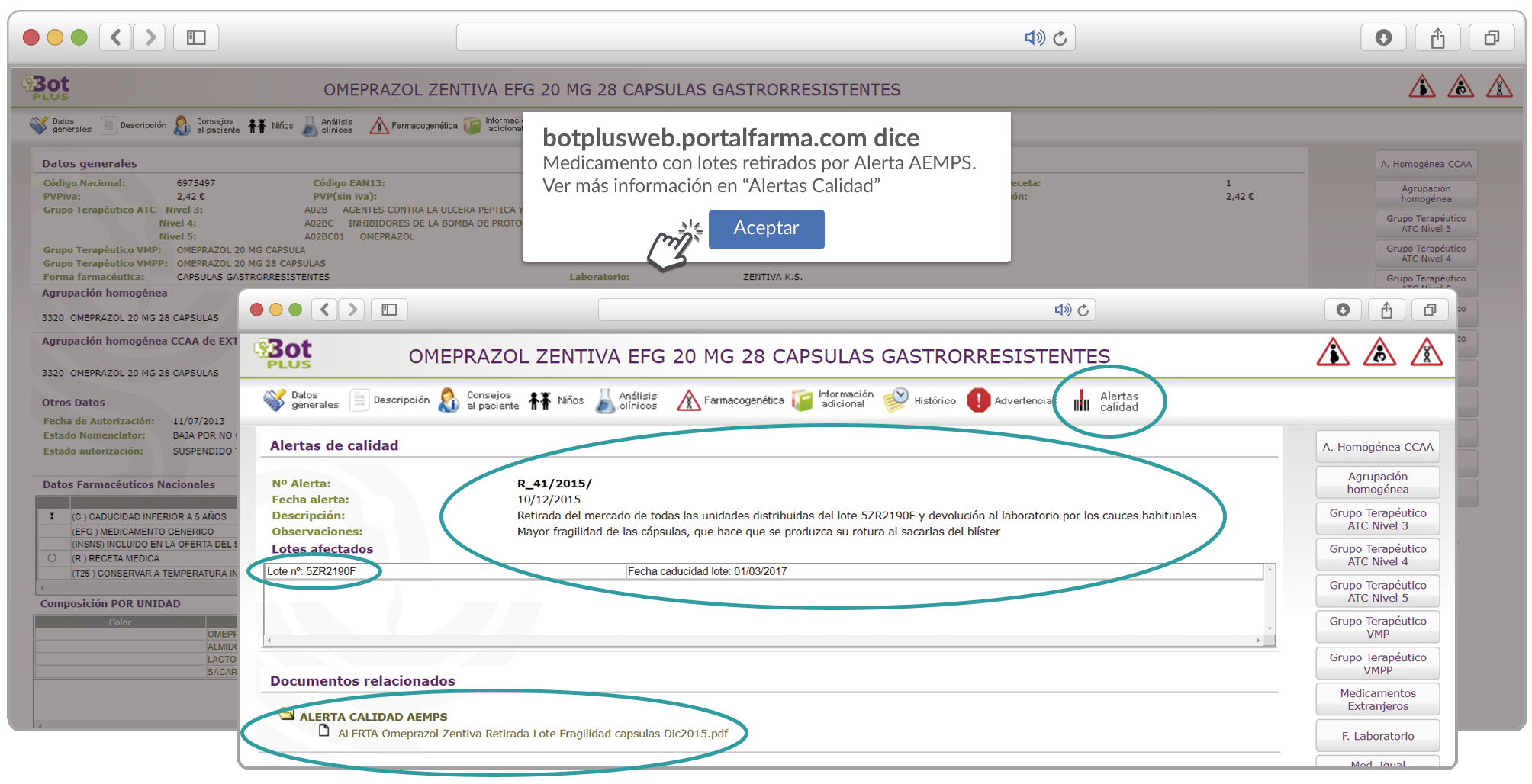
De forma interesante, dicha información se puede explotar a través de la Búsqueda Libre de Bot PLUS para obtener listados de todos los medicamentos afectados por alertas de calidad que implica la retirada (o también la inmovilización) de sus lotes en un momento dado.
Esta codificación de los lotes retirados es una información puesta a disposición de todos los usuarios, con el objetivo de ofrecer una nueva información capaz de integrarse con otros sistemas de información y mejorar la gestión e identificación de estos medicamentos, en los que la labor asistencial y de control del farmacéutico es fundamental.
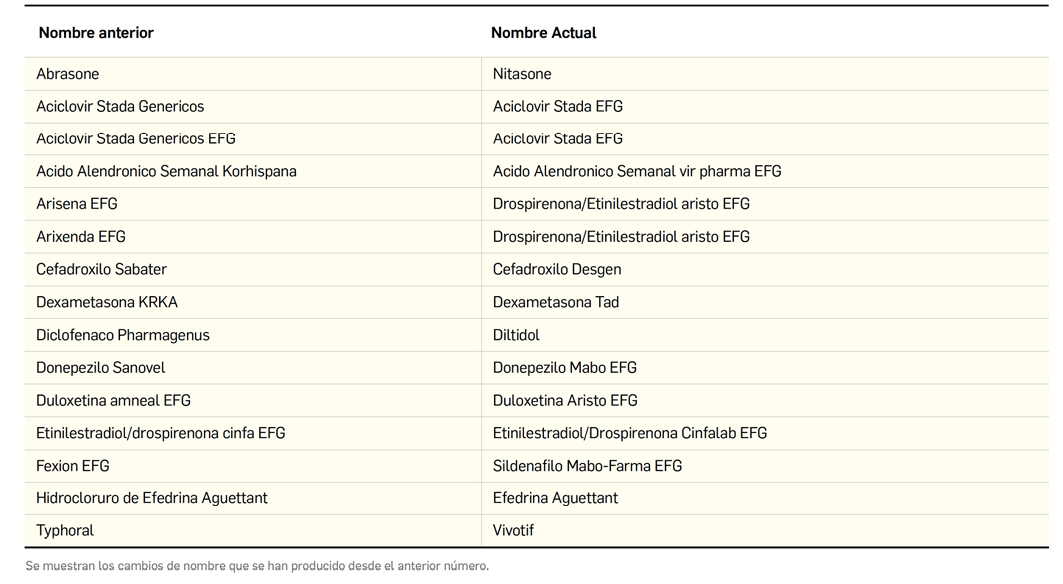
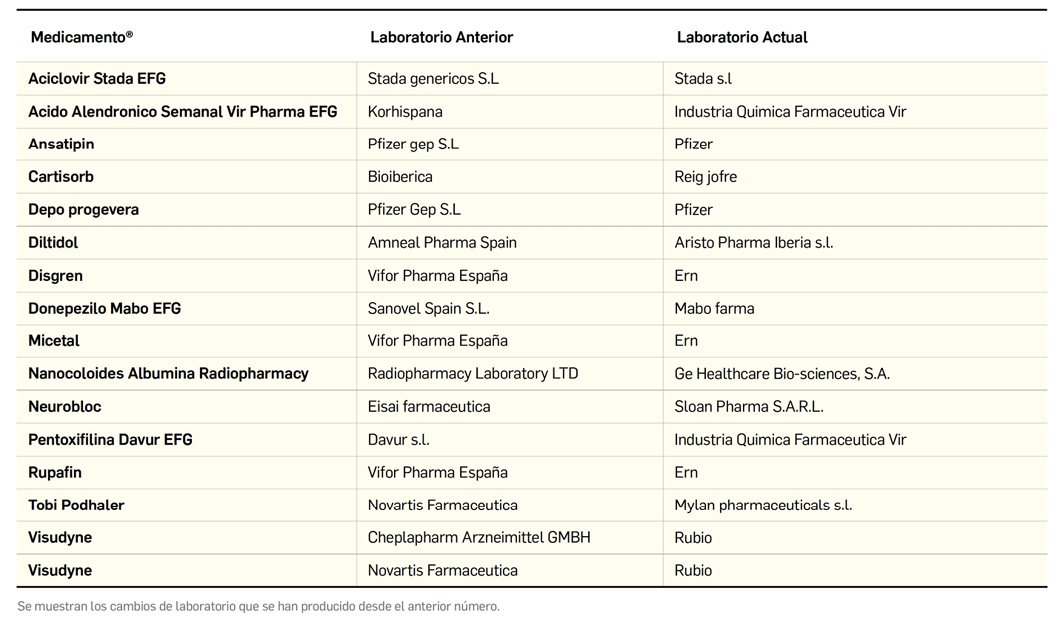
Además de la información que se incluye en los listados mensuales publicados en PAM, en Bot PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
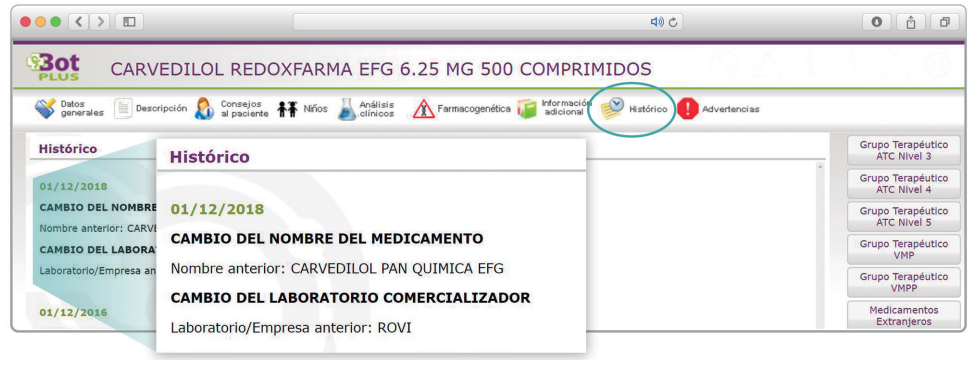
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre.
Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.
Además de la información existente en Histórico, se permite la explotación de la información incluida en Bot PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de Bot PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.

Es importante indicar que lo que se valora es el grado de innovación. Todos los medicamentos, innovadores o no, tienen utilidad terapéutica. Su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, seguridad, calidad y condiciones de uso (información incluida en la ficha técnica –sumario de características– y el prospecto del medicamento). La valoración que aquí se realiza se refiere a la incorporación, en un grado u otro, de algún elemento innovador comparado con otros medicamentos previamente autorizados para indicaciones terapéuticas iguales o similares o, en su caso, cubriendo la ausencia de éstas.
Además, debe considerarse que dicha evaluación coincide con la comercialización inicial del medicamento. Se trata, por tanto, de una valoración provisional de la innovación en función de la evidencia clínica disponible hasta el momento, lo que no prejuzga, en ningún caso, la existencia posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
El resultado se clasifica en tres niveles en función de la relevancia de la(s) innovación(es) del nuevo medicamento y siempre con el arsenal terapéutico disponible en España como referencia:
– SIN INNOVACIÓN (*). No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas.
– INNOVACIÓN MODERADA (**). Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar.
– INNOVACIÓN IMPORTANTE (***). Aportación sustancial a la terapéutica estándar.
También se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
– Evidencia clínica: obtenida de estudios controlados, diseñados y desarrollados para demostrar la eficacia y seguridad del nuevo medicamento y que demuestran de forma fehaciente lo que puede ser un avance o mejora sobre la terapia estándar, en el que caso de que exista.
– Plausibilidad científica (potencialidad): existencia de aspectos que teórica y racionalmente podrían mejorar la terapéutica actual, pero que no han sido demostrados con ensayos clínicos, por motivos éticos o imposibilidad de realizarlos en el momento de comercializar el medicamento: perfil de interacciones, mecanismos nuevos que inauguran vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinaciones con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia económica, etc.
El rigor de los datos contrastados en ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación; las potencialidades, en cambio, solo se valoran de forma accesoria, como aspectos complementarios de la valoración. En ningún caso, un medicamento es valorado como innovación importante en función de sus ventajas potenciales si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. La fundamental y determinante es la novedad clínica.
Binimetinib es un inhibidor –reversible y no competitivo con ATP– de las proteínas cinasas extracelulares activada por mitógenos (MEK) 1 y 2; por su parte, encorafenib es inhibidor –competitivo con ATP– potente y selectivo de las proteínas cinasas RAF. Su coadministración ejerce un efecto sinérgico que resulta en un mayor grado mayor de inhibición de la ruta de señalización RAS/RAF/MEK/ERK y, en consecuencia, en una menor proliferación de las células tumorales con mutación en la serina-treonina cinasa BRAF, que se identifica hasta en el 50% de los melanomas cutáneos. Por ello, ambos medicamentos han sido autorizados en combinación para el tratamiento –por vía oral– del melanoma no resecable o metastásico con mutación BRAF V600 en pacientes adultos.
Los datos clínicos derivan fundamentalmente de un estudio pivotal de fase 3 –COLUMBUS (N=577)–, abierto y con comparador activo, que en una primera parte demostró que la combinación reduce el riesgo de progresión de la enfermedad en un 46% y duplica la mediana de la supervivencia libre de progresión (SLP) en comparación con vemurafenib en monoterapia (14,9 vs. 7,3 meses); esa eficacia se verificó en todos los subgrupos de pacientes, a excepción de aquellos con metástasis cerebrales. También indujo una tasa de respuesta global (TRG, 63% vs. 40%) y una mediana de supervivencia global (SG) significativamente superiores al control (33,6 vs. 16,9 meses). En comparación con encorafenib en monoterapia, los resultados de SLP y SG fueron favorables para la combinación pero no se alcanzó significación estadística. Una segunda parte del estudio demostró que binimetinib contribuye significativamente en la eficacia de la combinación, pues ésta prolongó la mediana de SLP en 3,7 meses adicionales frente a encorafenib en monoterapia (12,9 vs. 9,2 meses), elevando la TRG en más de 15 puntos porcentuales (66% vs. 50%).
Por otro lado, el perfil toxicológico de la combinación es importante (equiparable al de otros agentes inhibidores de proteína cinasa y sus combinaciones empleadas en el tratamiento de melanoma avanzado), aunque se puede manejar con cambios posológicos. La mayoría de los eventos adversos fueron de grado ≥3 (58%), con tasas elevadas de toxicidad gastrointestinal –náuseas, diarrea, vómitos y estreñimiento–, fatiga y elevación de creatinín-cinasa; en términos cualitativos, destacan los trastornos oculares, los eventos hemorrágicos, la eritrodisestesis palmoplantar y la hipertensión. No obstante, parece que la combinación se tolera mejor que la monoterapia con encorafenib (tasas de abandono de 12% vs. 18% y de ajuste/interrupción posológica de 52% vs. 71%), que se caracteriza por una mayor toxicidad neurológica y, sobre todo, cutánea (con alta frecuencia de alopecia o hiperqueratosis, entre otras reacciones adversas).
No se dispone a día de hoy de comparaciones directas ni indirectas con las dos combinaciones recomendadas de inhibidores de RAF y MEK cinasas que han demostrado beneficio de supervivencia en melanoma inoperable/metastásico (trametinib-dabrafenib y cobimetinib-vemurafenib); parece que la eficacia de binimetinib-encorafenib sería similar, si bien la variabilidad en las poblaciones estudiadas impide extraer conclusiones. Sin innovación en cuanto a mecanismo de acción, esta asociación de nuevos fármacos viene a posicionarse como una alternativa terapéutica más en el manejo del melanoma avanzado con mutaciones de BRAF, cuadros en que se puede optar tanto por este tipo de terapias dirigidas como por la inmunoterapia anti-PD-1.
Los melanomas son tumores cutáneos malignos que se originan a partir de los melanocitos, aunque estos también pueden dar lugar a crecimientos benignos (no cancerosos), como son los lunares o nevus melanocíticos. El melanoma es indudablemente la forma de cáncer cutáneo más peligrosa, por su facilidad para diseminarse por otros tejidos generando metástasis en diversos órganos distales (cerebro, hígado, pulmones, etc.), aunque si se diagnostica con la antelación necesaria, puede ser eficazmente erradicado.
La prevalencia del melanoma tiene una gran variabilidad geográfica. Es uno de los 10 tumores malignos más frecuentes en el mundo occidental, siendo raro en la mayoría de los países de África, Asia y Sudamérica. La mayor incidencia se registra en países con alta irradiación ultravioleta (UV) y una población predominante de piel clara, como Australia y Nueva Zelanda, con 60 casos anuales por cada por 100.000 habitantes. En general, la incidencia del melanoma en todo el mundo aumenta a medida que nos acercamos al Ecuador, pero no así en Europa, donde es más frecuente en países nórdicos. En las últimas décadas, su importancia ha crecido notablemente hasta convertirse en uno de los tumores malignos con mayor incidencia en la población blanca. Se estima que cada 10 o 20 años se duplica su número.
España tiene una de las tasas de incidencia y mortalidad por melanoma más bajas de los países desarrollados: representa aproximadamente el 1,3% y el 2,5% de los tumores malignos en hombres y mujeres, respectivamente, mientras que actualmente la tasa estandarizada en todo el mundo es del 2,4% y 4,9%, respectivamente. Se ha estimado una tasa global de 8-10 nuevos casos/100.000 personas-año para el melanoma maligno, con máxima incidencia en Marbella (17,5) y mínima en Zaragoza (3,6). Sin embargo, es una de las patologías de mayor crecimiento en nuestro país, con aumentos porcentuales de >180% en la tasa de incidencia en hombres y >205% en las mujeres. Ese crecimiento de la incidencia afecta a todas las edades y es solo superado por los cánceres de hígado y de tiroides; sin embargo, la mortalidad permanece estable, probablemente en relación con la mejoría diagnóstica y la precocidad quirúrgica.
En el registro nacional de la Academia Española de Dermatología y Venereología (periodo 1998-2011) se observó que en nuestro país el melanoma es más común en las mujeres (57,2%), con una edad media al diagnóstico de 55 años en las mujeres y 57 años en los hombres, siendo el subtipo histológico más frecuente el de extensión superficial (60%). Las últimas predicciones divulgadas sobre la situación epidemiológica en España, en base a datos de 2018, apuntan a que en 2019 se diagnosticarán unos 6.200 casos nuevos de melanoma, de los cuales aproximadamente 2.500 serán en hombres y 3.700 en mujeres. En 2018, algo más de 18.000 personas vivían con melanoma en España, lo que supone el 2,4% del total de la prevalencia (a los 5 años del diagnóstico) de cáncer. En el año 2017, se notificaron un total de 992 muertes por melanoma (566 varones y 426 mujeres) (SEOM, 2019).
Como se ha indicado, los melanomas derivan de los melanocitos, fundamentalmente de aquellos localizados en la unión dermoepidérmica; la mayoría de los casos (75-80%) se originan de novo, es decir, sobre piel aparentemente normal y sin encontrarse un nevus melanocítico asociado. En general, es más común en partes descubiertas de la piel y en personas inmunosuprimidas. Aunque más del 95% de los casos de melanoma tienen localización cutánea, no se considera un cáncer exclusivamente cutáneo ya que puede aparecer en ojo, mucosas, tracto gastrointestinal, tracto urogenital, meninges y nódulos linfáticos. A pesar de ser el tipo de cáncer de piel más popularmente conocido, no se trata del tumor cutáneo más común (representa solo el 4% de las lesiones cancerígenas cutáneas). En cualquier caso, es el tumor maligno cutáneo más importante en razón de su pronóstico, siendo responsable de la mayoría (80%) de las muertes asociadas a cánceres cutáneos. Si se tiene en cuenta su alta letalidad y el hecho de que aproximadamente un tercio de los pacientes se diagnostican en edades inferiores a los 40 años, nos encontramos con uno de los tumores que ocasiona mayor número de años de vida perdidos y costes económicos asociados.
El melanoma se presenta predominantemente en adultos y más de 50% de los casos aparecen en áreas de la piel que son aparentemente normales. Entre los primeros signos en el nevo que indican cambios malignos están una coloración más oscura o variable, picazón, un aumento en el tamaño o el surgimiento de formaciones satélites; la ulceración o hemorragia son signos tardíos. El melanoma en mujeres se presenta con mayor frecuencia en las extremidades, y en hombres generalmente se presenta en el tronco, o en la cabeza y cuello, pero puede surgir en cualquier sitio de la superficie cutánea. La lesión típica es una mancha, generalmente asimétrica, mayor de 6 mm de diámetro, cuya coloración varía desde el marrón claro hasta el negro intenso, y generalmente irregular, con bordes dentados. Estas 4 características clínicas constituyen la denominada regla ABCD del melanoma (A: asimetría; B: bordes irregulares; C: color intenso; y
D: diámetro mayor de 6 mm). Sin embargo, tales características no constituyen criterios diagnósticos absolutos, pues también pueden observarse en lesiones melanocíticas benignas así como hay melanomas que no las cumplen.
Clínicamente, se distinguen cuatro tipos de melanoma:
En la génesis del melanoma intervienen factores genéticos y adquiridos. De entre estos, la exposición a la luz UV parece el de mayor importancia, singularmente si es esporádica e intensa, aunque no todos los melanomas están relacionados con el sol. Aparecen más frecuentemente en fototipos I y II (se ponen morenos con dificultad pero se queman fácilmente y se suele acompañar de ojos y pelo claros). La existencia de antecedentes personales o familiares de melanoma o de quemaduras solares en la niñez, un número elevado de nevos, y la presencia de nevos congénitos grandes son considerados factores de riesgo.
En cualquier caso, la genética tiene una relevancia especial en el melanoma, ya que las mutaciones típicamente asociadas al melanoma están presentes en el 10-40% de las familias con una tasa elevada de melanoma. En este sentido, más del 65% de los melanomas contienen mutaciones activadoras de la vía RAS/RAF/MEK/ERK, entre las que las mutaciones en la cinasa BRAF se llegan a identificar en el 40-50% de los melanomas cutáneos, en particular en la posición V600 (sustitución de valina por otro aminoácido en dicha posición de la cadena proteica de la cinasa); entre el 74% y el 90% de las mutaciones V600 detectadas en clínica son V600E (valina por ácido glutámico), seguidas en frecuencia por la V600K (15-25%); muchos menos comunes son las V600R, D, G, M, A y WT. Otro 10-15% tienen la mutación NRAS.
Por otro lado, los propios antecedentes personales son un factor de riesgo asociado al melanoma, ya que una persona que lo ha padecido anteriormente tiene un mayor riesgo de padecer otro melanoma. Se estima que el 5% de las personas con melanoma padecerán un segundo melanoma en algún momento de sus vidas.
En cuanto al diagnóstico, se debe sospechar clínicamente de melanoma en base a las características macroscópicas de la lesión cutánea y en el estudio de sus características dermatoscópicas1. No obstante, la confirmación del diagnóstico se realiza mediante el estudio histopatológico e inmunohistoquímico de la lesión, en el cual se especifica, además del subtipo histológico, la profundidad en la piel (medida en milímetros, es el llamado nivel de Breslow) y la presencia o ausencia de otras características que pueden influir en el pronóstico (ulceración, mitosis, regresión, infiltración linfocitaria, invasión linfática o neurotropismo). Dependiendo del índice de Breslow y del resto de las características del tumor se realizan pruebas de imagen (tomografía por emisión de positrones y tomografía axial computarizada) y la técnica del ganglio centinela (GC) para determinar la estadificación y el pronóstico del melanoma. Ésta última consiste en detectar mediante gamma-cámara el primer ganglio de drenaje linfático inyectando en la misma localización del melanoma un trazador radioactivo (tecnecio 99): si este primer ganglio de drenaje resulta afectado por células de melanoma, se procederá a realizar linfadenectomía del territorio.
El abordaje terapéutico del melanoma maligno depende fundamentalmente de su estadio al momento del diagnóstico, debiendo ser, de forma óptima, lo más precoz posible e individualizado de acuerdo con el tipo de tumor, el tipo de paciente y la experiencia clínica.
El tratamiento de elección de la enfermedad localizada cutánea, que debe buscar la curación del proceso, es la extirpación quirúrgica completa dejando márgenes de piel libre de enfermedad alrededor del tumor que dependen de la profundidad (nivel de Breslow) del melanoma al momento del diagnóstico: melanoma in situ, 0,5-1 cm; para melanomas de 1-2 cm de grosor, 1-2 cm de margen; para melanomas >2 cm de grosor, 2 cm de margen. La mayoría de los pacientes con melanoma se diagnostican en estadios tempranos con la enfermedad localizada y la extirpación quirúrgica puede ser curativa, empleando por lo general la técnica del ganglio centinela.
Cuando la lesión se localiza en sitios poco reconocibles (región plantar, subungueal, cuero cabelludo, mucosas) o cuando carece de pigmentación (melanoma amelanótico), el diagnóstico puede retrasarse y empeorar el pronóstico. Algunos casos –se estima una tasa cercana al 2% en España– se presentan con enfermedad localmente avanzada, con afectación de los ganglios linfáticos regionales o con metástasis por melanoma en órganos distantes (estadios III-IV), bien de inicio o porque progresan tras el tratamiento inicial; hasta el 20% de los pacientes diagnosticados de melanoma desarrollan metástasis, y la regresión espontánea ha sido reportada con una incidencia menor del 1%.
Esos casos de pacientes con melanoma metastásico o irresecable (no operable) quirúrgicamente tienen un pronóstico poco favorable, con promedios de supervivencia cercanos a los 8 meses sin tratamiento (6-9 meses) y una tasa de supervivencia al cabo de 1 año desde el diagnóstico del 25%, que desciende al 15% a los cinco años. En esos supuestos clínicos, es necesaria la administración de tratamientos farmacológicos sistémicos adyuvantes o de tratamiento de la enfermedad metastásica; la radioterapia también puede utilizarse como terapia complementaria. Hasta hace poco, las opciones para el manejo terapéutico de los pacientes con melanoma avanzado eran muy escasas, pero su tratamiento ha experimentado notables avances en los últimos 5-7 años, pasando de ser una enfermedad en la que prácticamente la única opción terapéutica era la quimioterapia paliativa (con una expectativa de vida limitada) a una situación en que las opciones terapéuticas actuales son múltiples y se asocian a una importante prolongación de la supervivencia. Todavía se carece, no obstante, de un tratamiento curativo de este tipo de tumores.
El tratamiento “clásico” del melanoma metastásico ha consistido en el empleo de quimioterapia, y más concretamente, de dacarbazina, considerado durante mucho tiempo como el fármaco de mayor eficacia en monoterapia, situándose sus tasas de respuesta en torno al 20% (aproximadamente un 5% son remisiones completas), con una mediana de duración de las respuestas de 4-6 meses; los principales efectos secundarios son náuseas y vómitos. Por su parte, la temozolomida es semejante a la dacarbazina, pero es capaz de atravesar la barrera hematoencefálica y puede ser absorbida por vía oral; por ello, se ha utilizado preferentemente como tratamiento de las metástasis cerebrales del melanoma. Se pueden utilizar, asimismo, diversos tratamientos combinados de quimioterapia, asociando dacarbazina junto con otros agentes citotóxicos, como cisplatino, vinblastina, carmustina o tamoxifeno.
A grandes rasgos, para el tratamiento del melanoma maligno avanzado irresecable o metastásico, uno de los criterios actuales de selección de tratamiento es la presencia de mutaciones en el gen BRAF V600 (sobre el 40-50% de casos), que permiten el empleo de fármacos inhibidores específicos que bloquean la señalización de esta vía oncogénica. Las principales guías clínicas actualmente recomiendan como primera línea de tratamiento una de las siguientes opciones (Fernández-Moriano, 2019):
Los datos disponibles hasta el momento apuntan a que la inmunoterapia podría conducir a tasas de supervivencia libre de progresión (SLP) superiores en la población global de pacientes con melanoma avanzado inoperable: tasas de SLP a 4 años del 31-36% con pembrolizumab y del 32% a 3 años con nivolumab, frente a tasas de SLP a 4-5 años del 13% con terapia dirigida con dabrafenib más trametinib (y comportamiento posiblemente similar para vemurafenib+cobimetinib). No obstante, se debe subrayar que las poblaciones de tratamiento son diferentes, pues la terapia dirigida solo se indica en pacientes que presentan mutación de BRAF V600 (que, por lo general, tienen peor pronóstico) mientras que los ensayos clínicos con inmunoterapia incluyen tanto pacientes mutados como no mutados. En definitiva, en base a la evidencia disponible, es difícil priorizar un tipo de terapia sobre la otra, pero hay estudios clínicos en marcha que darán respuesta a esta cuestión.
Debido al desarrollo de resistencias, la mayor parte de pacientes suele recibir ambos tratamientos de forma secuencial. Sin embargo, no está muy claro aún cuál es la secuencia óptima de estos tratamientos, pues no se dispone de estudios controlados comparativos (la mayor parte de la información proviene de análisis de subgrupos de diferentes ensayos y de pequeños estudios fase II). Se considera que la inmunoterapia con anti-PD1 puede ser adecuada en pacientes que no necesitan una rápida respuesta y parece que podría ser preferente su uso en el inicio del tratamiento del melanoma, pues un menor número de pacientes podría requerir cambio terapéutico.
Binimetinib es un inhibidor reversible, de molécula pequeña y no competitivo con el ATP, de las proteínas cinasas extracelulares activadas por mitógenos (MEK) 1 y 2, mientras que encorafenib es una molécula pequeña competitiva con ATP que inhibe potente y selectivamente las proteínas cinasas RAF. En base a esos mecanismos, ambos medicamentos –activos por vía oral– han sido autorizados en combinación para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con mutación BRAF V600.
Ambas proteínas cinasas (MEK-1, MEK-2 y RAF) forman parte de la vía de las cinasas reguladas por señales extracelulares (ERK, extracellular signal-regulated kinases), que promueve la proliferación celular. Concretamente, las proteínas MEK son reguladores retrógrados de dicha vía. Cabe destacar que en el melanoma y otros tipos de tumores esta ruta de señalización está con frecuencia sobre-activada por formas mutadas de la serina/treonina cinasa BRAF, que activan a las MEK y están relacionadas con la proliferación celular incluso en ausencia de factores de crecimiento. De hecho, se ha descrito que más del 65% de los melanomas contienen mutaciones activadoras de la vía extracelular RAS/RAF/MEK/ERK, entre las que las mutaciones en la cinasa BRAF se llegan a identificar en el 40-50% de los melanomas cutáneos, en particular en la posición V600 (sustitución de valina por otro aminoácido en dicha posición de la cadena proteica de la cinasa); entre el 74% y el 90% de las mutaciones V600 detectadas en clínica son V600E (valina por ácido glutámico), seguidas en frecuencia por la V600K (15-25%); muchos menos comunes son las V600R, D, G, M, A y WT.
Binimetinib ha demostrado la capacidad de inhibir la activación de las MEK-1 y MEK-2 (mitogen-activated extracellular signal regulated kinase 1 y 2) por el BRAF, con una concentración inhibitoria media (CI50) en el rango de 12 a 46 nM. Este efecto inhibidor de cinasas –que se traduce en el bloqueo de los procesos de fosforilación proteicos que median multitud de funciones biológicas– se ha asociado con la inhibición in vitro del crecimiento de líneas celulares de melanoma con mutación BRAF V600 y con los efectos antitumorales que exhibe el fármaco en modelos animales con dicha mutación.
Por su parte, encorafenib ha demostrado una inhibición más potente de las proteínas RAF: su CI50 frente a BRAFV600E, BRAF y CRAF se ha estimado en 0,35, 0,47 y 0,30 nM, respectivamente; se trata, además, de una inhibición prolongada de la vía RAF/MEK/ERK, ya que su semivida de disociación se estima superior a 30 h. En estudios preclínicos, se ha demostrado que encorafenib ejerce ese efecto farmacológico en diversas en células tumorales que expresan formas mutadas de BRAF cinasa (V600E, D y K), inhibiendo in vitro e in vivo el crecimiento de las células de melanoma que expresan esas proteínas mutantes. Sin embargo, parece que encorafenib carece prácticamente de actividad sobre otras tirosina y serina/treonina cinasas implicadas en los procesos de señalización celular, y no inhibe la actividad de las proteínas de la vía RAF/MEK/ERK en células que no tienen el gen BRAF mutado (es decir, cuando las proteínas son normales), lo cual permite descartar un efecto antiproliferativo en las células sanas del organismo (EMA, 2018).
Según estos mecanismos, se entiende que la acción conjunta de binimetinib con encorafenib provocará un efecto sinérgico y un grado mayor de inhibición –en dos puntos diferentes– de la ruta de señalización intracelular y de reducción de la proliferación de las células tumorales con mutación BRAF. De hecho, se ha demostrado que la combinación de ambos fármacos impide la aparición de resistencias al tratamiento en xenoingertos en modelos animales de melanoma humano con mutación BRAF V600E. Ambos fármacos carecen prácticamente de actividad sobre otras proteínas cinasas implicadas en los procesos normales de señalización celular.
Binimetinib y encorafenib están estrechamente relacionados estructural y farmacológicamente con otros miembros de la serie de inhibidores de proteína cinasas (Figura 1), en la que se encuadran un gran número de principios activos comercializados en España. Como otros miembros de la serie, son el resultado de la optimización funcional mediante modelización molecular a partir de una serie de 2-fenilaminopirimidinas, de donde surgió el imatinib, cabeza de serie del grupo.
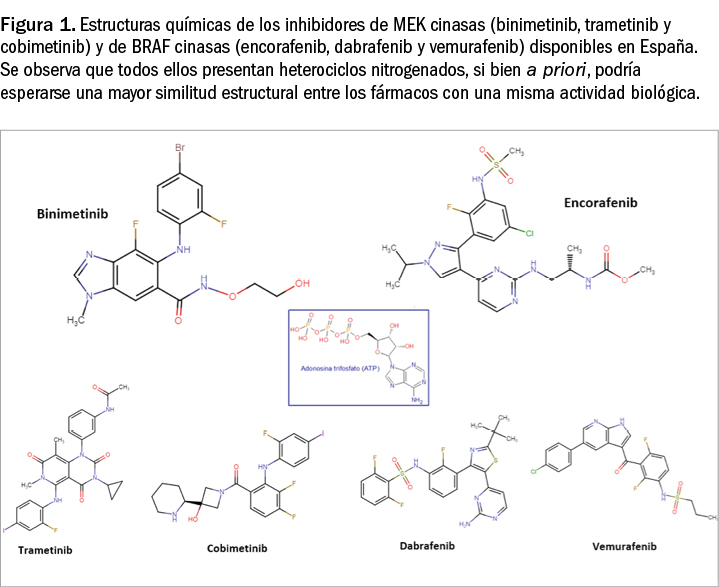
Aunque se aprecia una diversidad estructural importante en el grupo, todos ellos presentan heterociclos derivados azólicos y guardan –en mayor o menor grado– una familiaridad química con la molécula de ATP (o, en su caso, con la de GTP, como sucede en las cinasas MAPK), con la cual algunos de los principios activos compiten para provocar el bloqueo de la cinasa correspondiente. Se han desarrollado modelos moleculares de relación estructura-actividad para este grupo de sustancias y, en todos los casos, las interacciones estéricas y electrostáticas han demostrado ser determinantes para el efecto inhibitorio sobre la tirosina cinasa.
Binimetinib tiene el nombre químico 5-[(4-bromo-2-fluorofenil)amino]-4-fluoro-N-(2-hidroxietoxi)-1-metil-1H-benzimidazol-6-carboxamida, que se corresponde con la fórmula C17H15BrF2N4O3 y un peso molecular de 441,23 g/mol. Se trata de una molécula sin centros quirales que se presenta como un polvo blanco o amarillo pálido.
Encorafenib, por su parte, tienen como nombre químico el metil N-{(2S)-1-[(4-{3-[5-cloro-2-fluoro-3-(metanosulfonamida)fenil]-1-(propan-2-il)-1H-pirazol-4-il}pirimidin-2-il)amino]propan-2-il}carbamato, que se corresponde con la fórmula molecular C22H27ClFN7O4S y el peso molecular de 540 g/mol. Se trata de una molécula que exhibe estereoisomería por la presencia de un único centro quiral, se presenta como un polimorfo único y en forma de polvo blanquecino no higroscópico; su solubilidad al pH normal del estómago y su permeabilidad aparente son elevadas.
La eficacia y la seguridad clínicas de binimetinib en combinación con encorafenib han sido adecuadamente contrastadas en la indicación autorizada mediante un ensayo clínico pivotal de fase 3 –confirmatorio de eficacia y seguridad– (estudio COLUMBUS o CMEK162B2301), aleatorizado, multicéntrico, abierto y con control activo, realizado en pacientes adultos con melanoma irresecable en estadio IIIB o IIIC (avanzado) o IV (metastásico) –según la clasificación del American Joint Committee on Cancer– y con confirmación de la mutación BRAF V600E o K.
El estudio se dividió en dos partes. En la Parte 1, se comparó la farmacodinamia de la combinación binimetinib+encorafenib (combo) frente a vemurafenib y frente a encorafenib, ambos en monoterapia. Para ello, se aleatorizaron un total de 577 pacientes (1:1:1) a recibir el combo con 45 mg/12 h de binimetinib y 450 mg/24 h de encorafenib (N=192), 0 vemurafenib 960 mg/12 h (N=191) o encorafenib 300 mg/24 h (N=194), continuándose el tratamiento hasta progresión de la enfermedad o aparición de toxicidad inaceptable. Como variable principal de eficacia se evaluó por un comité revisor independiente la supervivencia libre de progresión (SLP) del combo frente a vemurafenib, siendo la comparación de SLP del combo vs. encorafenib en monoterapia una variable secundaria; también se estimaron la supervivencia global (SG), la tasa de respuesta global (TRG), la duración de la respuesta (DR) y la tasa de control de la enfermedad, evaluadas por el comité de revisión independiente y por los investigadores.
Los principales criterios de inclusión fueron: edad ≥18 años, puntuación ECOG 0-1 y ausencia de tratamientos previos con otros inhibidores de BRAF o MEK; sin embargo, se excluyeron pacientes con melanoma uveal o mucoso, con patología cardíaca, metástasis en el sistema nervioso central, patologías infecciosas o con riesgo de oclusión retiniana. Las características demográficas y de enfermedad basales estaban bastante balanceadas entre los 3 brazos de tratamiento. En global, la mediana de edad fue de 56 años, un 58% eran varones, el 90% eran caucásicos y el 72% tenían un estado funcional muy bueno (ECOG 0); además, el 95% presentaban enfermedad metastásica en estadio IV (un 64% en estadio IVM1c), el 45% tenían afectación de ≥3 órganos (3,5% con metástasis cerebrales), el 27% presentaban valores séricos elevados de lactato deshidrogenasa (LDH) y un 5% de pacientes había recibido 1 línea de inmunoterapia previa (monoterapia con anti PD-1/PD-L1 o en combinación con ipilimumab).
Con una mediana de exposición al tratamiento de 11,7, 6,2 y 7,1 meses para los pacientes tratados con el combo, con vemurafenib y con encorafenib, respectivamente, los resultados publicados (Dummer et al., 2018a; Dummer et al., 2018b) demuestran que, en un primer punto de corte, la combinación de los nuevos fármacos permitía alcanzar una mediana de SLP –evaluada por comité ciego independiente– de 14,9 meses (IC95% 11,0-18,5), mientras que la SLP con vemurafenib y con encorafenib en monoterapia se quedaba en 7,3 (IC95% 5,6-8,2) y 9,6 meses (IC95% 7,5-14,8), respectivamente. Las comparaciones entre brazos de tratamiento confirman que la mejoría en SLP con la combinación es estadísticamente significativa en comparación con vemurafenib (HR: 0,54; IC95% 0,41-0,71; p<0,001), pero no alcanza esa significación estadística cuando se compara con la monoterapia con encorafenib (HR: 0,75; IC95% 0,56-1,00; p= 0,051), la cual también fue superior a vemurafenib en términos de SLP (HR: 0,68; IC95% 0,52-0,90; p: 0,007). Tanto éstos como otros resultados de eficacia basados en la evaluación de los investigadores coincidieron y respaldan los del análisis central independiente.
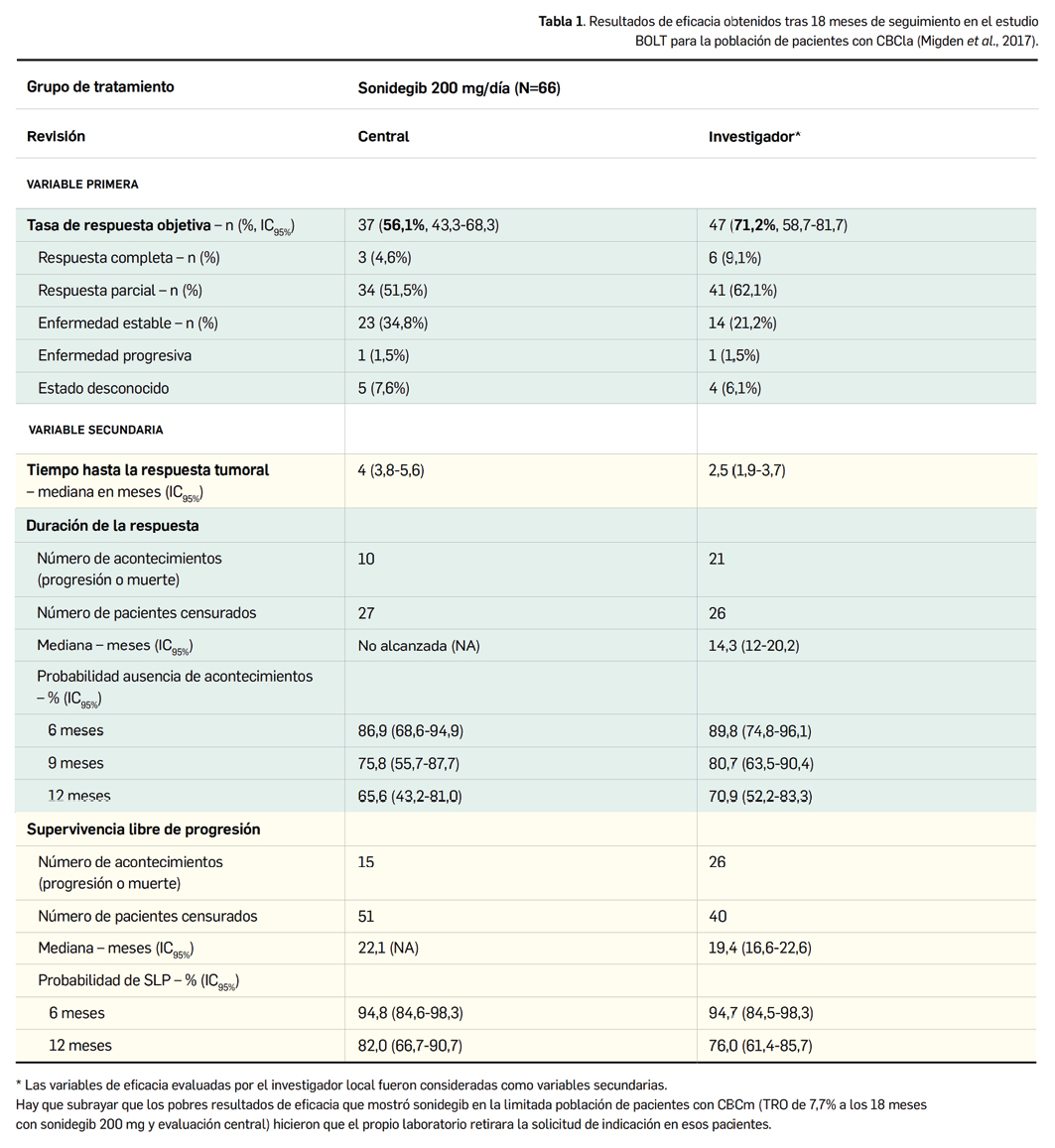
Cabe destacar que el beneficio en SLP de la combinación en estudio se verificó en todos los subgrupos de pacientes analizados (en función de tipo de mutación tumoral, sexo, región geográfica, edad, estadio, etc.), salvo para aquellos con metástasis cerebrales, aunque la limitación del número de pacientes de ese subgrupo (N= 12) impide extraer conclusiones al respecto. Las consideraciones aquí realizadas se refrendan con los datos del ensayo clínico derivados de un punto de corte un año posterior. Los resultados de eficacia referentes a las variables secundarias se reflejan en la siguiente tabla (Tabla 1); los datos de SG derivan de la actualización de datos tras el segundo punto de corte.

Adicionalmente, en esta primera parte se investigó el efecto de los tratamientos sobre la calidad de vida relacionada con la salud (los resultados percibidos por los pacientes), mediante tres cuestionarios validados, como son el de Evaluación Funcional para el Tratamiento del Cáncer – Melanoma (FACT-M), el de la European Organization for Research and Treatment of Cancer (EORTC QLQ-C30) y el cuestionario de 5 dimensiones y 3 niveles del Grupo EuroQoL (EQ-5D-5L). En los dos primeros, con el tratamiento experimental en combinación se observó un retraso significativo del tiempo hasta un deterioro del 10% de los pacientes tratados (no se alcanzó la mediana del tiempo hasta el deterioro vs. 22,1 meses con vemurafenib; HR: 0,46), en comparación con las monoterapias con encorafenib o vemurafenib. En el cuestionario EQ-5D-5L, los pacientes del brazo de la combinación no mostraron mejoría pero tampoco deterioro, que sí se puso de manifiesto en los pacientes bajo cualquiera de las monoterapias.
La Parte 2 del ensayo pivotal presentó un diseño diferente, enfocado a evaluar la contribución de binimetinib a la combinación de éste con encorafenib. Para ello, en este caso se administró el combo con una dosis de 45 mg/12 h de binimetinib más 300 mg/24 h de encorafenib, comparando su eficacia frente al brazo ampliado de encorafenib de la Parte 1, ahora a la dosis de 300 mg/24 h. Se aleatorizaron 344 pacientes: 258 fueron tratados con el combo y 86 con encorafenib en monoterapia. De nuevo, las características de los pacientes estaban equilibradas entre ambos brazos, si bien los pacientes del brazo de encorafenib presentaron un pronóstico peor comparando entre la Parte 1 y la 2 (mayor edad, estadios más avanzados y mayor elevación de LDH).
La combinación de los resultados de eficacia –según el análisis del comité independiente– de esta parte con los de la Parte 1 demostró que binimetinib contribuye notablemente a la prolongación de la SLP por la combinación: se alcanzó una mediana de SLP de 12,9 meses (IC95% 10,1-14,0) con la combinación de binimetinib más encorafenib, mientras que ésta se quedó en 9,2 meses (IC95% 7,4-11,0) con encorafenib en monoterapia (HR: 0,77; IC95% 0,61-0,97); si se consideran solo los pacientes de la Parte 2, la SLP para la monoterapia con encorafenib es de 7,4 meses (HR: 0,57; IC95% 0,41-0,78). Además, se puso de manifiesto que la combinación con binimetinib mejora los resultados de tasa de respuesta global (65,9% para la combinación vs. 50,4% para encorafenib en monoterapia), aunque sin beneficio en la duración de la respuesta (mediana de 12,7 meses con la combinación vs. 12,9 meses con encorafenib en monoterapia).
Se debe mencionar que un análisis post hoc de los resultados de eficacia de la nueva combinación a ambas dosis de encorafenib (450 vs. 300 mg/24 h) no halló diferencias notables en SLP, situándose la mediana en 14,9 meses con la dosis de 450 mg y en 12,9 meses con 300 mg, ni tampoco en la TRG objetiva (63% con 450 mg vs. 65,9% con 300 mg). En cambio, la duración de la respuesta fue mayor con la dosis de 450 mg de encorafenib (16,6 vs. 12,7 meses) y un análisis de regresión para los factores de estratificación demostró un mayor beneficio clínico con la combinación que incluye 450 mg/día de encorafenib (HR: 0,74; IC95% 0,56-0,98).
Con respecto a la seguridad clínica, el perfil toxicológico de binimetinib-encorafenib a las dosis autorizadas parece adecuadamente definido en base a los datos procedentes de 371 pacientes tratados en el ensayo pivotal y 2 estudios previos de fase 2 (mediana de exposición de 11,7 meses en el ensayo COLUMBUS), que se complementan con los datos obtenidos de pacientes tratados con la combinación que incluía encorafenib a 300 mg/día en la Parte 2 del ensayo pivotal (N= 258). En general, se documentaron eventos adversos en la práctica totalidad de pacientes tratados, si bien la combinación de binimetinib con encorafenib mostró una menor incidencia de eventos adversos graves (grado 3-4) que la monoterapia con encorafenib (57,8% vs. 66,1%), lo que se tradujo en una menor tasa de abandono por eventos adversos (11,7% vs. 18,0%) o de necesidad de ajuste/interrupción de tratamiento (52,2% vs. 71%).
Las reacciones adversas más frecuentes con la combinación (con encorafenib a 450 mg/día) fueron: náuseas (39,4%), diarrea (36,1%), fatiga (30,3%), vómitos (26,6%), elevación de creatinín-cinasa (24,8%), artralgia (24,8%) y estreñimiento (22,6%). Entre los eventos adversos graves que requirieron una modificación/interrupción de la pauta posológica fueron náuseas (6,6%), vómitos (5,8%), elevación de alanina aminotransferasa (5,5%) y reducción de la fracción de eyección cardiaca (5,1%). Cualitativamente, puede subrayarse la incidencia de uveítis (4,4%) y alteraciones visuales (21,5%), que cursaron con visión borrosa o pérdida de agudeza visual generalmente reversibles, desprendimiento de retina leve-moderado (29,6%), eventos hemorrágicos (17,9%), eritrodisestesis palmoplantar2 (6,2%) y aparición o agravamiento de la hipertensión (11,7%). No se hallaron diferencias notables de toxicidad en la comparación de las combinaciones de binimetinib con encorafenib a la dosis de 450 mg/día o de 300 mg/día.
En aquellos pacientes en que encorafenib se administró en monoterapia (300 mg/24 h; N= 217), destacaron, por su frecuencia, la incidencia de alopecia (56,2%), eritrodisestesia palmoplantar (51,6%), artralgia (42,9%), hiperqueratosis (41%), náuseas (37,8%), piel seca (31,3%), mialgia (29,5%) cefalea (28,1%), fatiga (27,6%) y vómitos (26,7%), y por su gravedad (grado 3-4), la incidencia de eritrodisestesia palmoplantar (12,4%), artralgia (9,2%) y mialgia (9,2%), que fueron también los que supusieron una mayor frecuencia de modificaciones/interrupciones del tratamiento (Gogas et al., 2019).
Binimetinib (un inhibidor reversible –y no competitivo con el ATP– de las proteínas cinasas extracelulares activada por mitógenos MEK-1 y MEK-2) y encorafenib (inhibidor potente y selectivo – competitivo con ATP– de las proteínas cinasas RAF) son dos nuevos agentes antineoplásicos activos por vía oral que, cuando se administran concomitantemente, ejercen un efecto farmacológico sinérgico que permite alcanzar un mayor grado mayor de inhibición –en dos puntos diferentes– de la ruta de señalización RAS/RAF/MEK/ERK y, en consecuencia, una reducción de la proliferación de las células tumorales con mutación en la serina-treonina cinasa BRAF, que se llega a identificar hasta en el 50% de los melanomas cutáneos.
En base a ello, ambos medicamentos han sido autorizados en combinación para el tratamiento en pacientes adultos del melanoma no resecable o metastásico con mutación BRAF V600. Los datos clínicos conducentes a dicha autorización derivan de un amplio estudio aleatorizado y abierto de fase 3 –COLUMBUS (N=577)– que, en dos partes diferenciadas, confirmó la eficacia y seguridad de la combinación de binimetinib con encorafenib en pacientes adultos con melanoma irresecable o metastásico (estadios III o IV).
En una primera parte, dicho ensayo comparó la eficacia de la combinación de los nuevos fármacos con el control activo vemurafenib (inhibidor de la RAF cinasa) en monoterapia3; también se comparó con el propio encorafenib en monoterapia. A las dosis autorizadas y en revisión por comité central independiente, la combinación binimetinib-encorafenib mostró la capacidad de duplicar –aumento de más de 7 meses– la mediana de la supervivencia libre de progresión (SLP) en comparación con vemurafenib (14,9 vs. 7,3 meses), lo que se tradujo en una reducción significativa de un 46% (HR: 0,54; p<0,0001) del riesgo de progresión de la enfermedad. Esa prolongación notable de SLP se verificó en todos los subgrupos analizados, a excepción de pacientes con metástasis cerebrales.
Los resultados de las variables secundarias de eficacia respaldan el beneficio clínico aportado por la combinación, que fue capaz de inducir una tasa de respuesta global (TRG) del 63% (con un 8% de respuestas completas –RC), superior en más de 20 puntos porcentuales a la alcanzada con vemurafenib (40%, 6% de RC) y mejorando la alcanzada con encorafenib en monoterapia (51%, 5% de RC). Sin grandes diferencias en la duración de la respuesta antitumoral entre los brazos de tratamiento, el resultado más relevante fue quizá el beneficio en términos de supervivencia global (SG), pues la combinación duplica la mediana (33,6 vs. 16,9 meses) y aumenta notablemente la tasa de supervivencia a los 24 meses (58% vs. 43%) frente a vemurafenib, lo que supone una reducción del 39% en el riesgo de muerte (HR: 0,61; p< 0,0001). Estos resultados se acompañaron de una mejoría en la calidad de vida percibida por los pacientes, con un retraso significativo del tiempo hasta un deterioro del 10% de pacientes. En la comparación con la monoterapia de encorafenib, la combinación obtuvo valores favorables de medianas SLP (14,9 vs. 9,6 meses) y de SG (33,6 vs. 23,5 meses) a pesar de no alcanzar significación estadística.
La parte 2 del estudio, con un diseño comparativo diferente, demostró que binimetinib contribuye significativamente en la eficacia de la combinación. De hecho, si bien la duración de la respuesta fue similar respecto al uso de encorafenib en monoterapia, la combinación binimetinib-encorafenib fue capaz de prolongar 3,7 meses adicionales la mediana de SLP (12,9 vs. 9,2 meses) y elevó la TRG en más de 15 puntos porcentuales (66% vs. 50%), aspectos ambos que evidencian una eficacia superior de la combinación frente a la monoterapia con encorafenib.
Desde el punto de vista de la seguridad, el perfil toxicológico de la combinación es importante, equiparable al de otros agentes inhibidores de proteína cinasa y de las combinaciones empleadas en el tratamiento de melanoma avanzado4 (e incluso similar al de la quimioterapia clásica), siendo la mayoría de los eventos adversos descritos de grado 3 o superior (58%). Predominan la toxicidad gastrointestinal –en forma de náuseas (39%, 7% graves), diarrea (36%), vómitos (27%, 6% graves) y estreñimiento (23%)–, la fatiga (30%) y la elevación de creatinín-cinasa (25%); en términos cualitativos, sobresalen los trastornos oculares (alteraciones visuales, desprendimiento de retina y uveítis), los eventos hemorrágicos, la eritrodisestesis palmoplantar y la hipertensión.
No obstante, parece que la combinación puede tolerarse mejor que la monoterapia con encorafenib (tasa de abandonos de 12% vs. 18% y de ajuste/interrupción posológica de 52% vs. 71%), que se caracteriza por una mayor toxicidad neurológica y, sobre todo, cutánea, según se manifiesta en mayores tasas de alopecia (70% vs. 16% con la combinación), hiperqueratosis (15% vs 51%), eritrodistesia palmoplantar (6,6% vs 56%), queratoma palmoplantar (9,7% vs 33%) y nevus melanocítico (2% vs 14%).
Por el momento, no se dispone de comparaciones directas (ni de comparaciones indirectas ajustadas) con las otras dos combinaciones recomendadas de inhibidores de RAF y MEK cinasas que hasta ahora han demostrado beneficio en términos de supervivencia en pacientes con melanoma inoperable/metastásico (trametinib-dabrafenib y cobimetinib-vemurafenib). Una visión global de los resultados de estudios clínicos podría sugerir que la eficacia de las tres combinaciones es similar: la mediana de SLP con binimetinib-encorafenib ha sido de 14,9 meses, de 12,3 meses con vemurafenib-cobimetinib y de 11-11,4 meses con dabrafenib-trametinib, y la mediana de SG se ha situado en 33,6, 22,3 y 25,6 meses, respectivamente; las TRG también son parecidas, todas en el rango del 63-70%. No obstante, la variabilidad en las poblaciones de estudio y la ausencia de ensayos comparativos impiden extraer conclusiones.
En resumen, la asociación binimetinib-encorafenib ha demostrado una prolongación significativa –duplicación– de la supervivencia respecto a la monoterapia con inhibidores de cinasas RAF, con un perfil de seguridad comparable al de otras combinaciones de terapia dirigida usadas en melanoma con similares resultados de eficacia (se aprecia una mayor toxicidad gastrointestinal y menor fotosensibilidad y pirexia). Ninguno de los dos nuevos fármacos representa una novedad en el plano mecanístico, al tratarse de inhibidores de proteínas cinasas que comparten mecanismo y diana farmacológica con otros fármacos ya autorizados en la misma indicación (binimetinib con trametinib o cobimetinib, y encorafenib con dabrafenib o vemurafenib).
Al menos dos meta-análisis (An et al., 2019; Zoratti et al., 2019) no han encontrado diferencias significativas en términos de supervivencia entre el uso de estas combinaciones de terapia dirigida frente a la inmunoterapia anti-PD-1 en pacientes con melanoma sin tratamiento previo, por lo que se puede concluir que la combinación aquí analizada representa una alternativa terapéutica más en el manejo de pacientes con melanoma con mutaciones de BRAF. Su uso estará limitado por el hecho de que aún no se puede recomendar esta nueva combinación en pacientes que han progresado al tratamiento con otros inhibidores de BRAF (criterio de exclusión) ni en cuadros con metástasis cerebrales, una de las principales causas de muerte en este tipo de pacientes. La elección del tratamiento de inhibidores BRAF/MEK o inmunoterapia debe ser individualizada, considerando la agresividad de la enfermedad y los perfiles de tolerabilidad de las opciones terapéuticas.


Sonidegib es un nuevo inhibidor de la ruta de señalización Hedgehog (Hh), capaz de unirse a la proteína transmembrana Smoothened e impedir la activación y localización nuclear de los factores de transcripción gli, inhibiendo así la proliferación, diferenciación y supervivencia de las células tumorales (mediadas por dicha ruta en hasta el 90% de los tumores cutáneos de células basales). En base a ello, el medicamento ha sido oficialmente autorizado para el tratamiento por vía oral de pacientes adultos con carcinoma basocelular localmente avanzado (CBCla) que no es susceptible a la cirugía curativa ni a radioterapia.
Los datos de eficacia y seguridad clínicas son limitados y derivan en su mayoría de un único ensayo pivotal de fase 2 (BOLT), abierto y no comparativo (N=230), por lo que las incertidumbres son notables. No obstante, la eficacia del fármaco parece clínicamente relevante. En pacientes con CBCla, sonidegib 200 mg/día ha demostrado inducir tasas de respuesta objetiva del 56,1% tras revisión central (71,2% en revisión por investigador). Las variables secundarias respaldan su eficacia, con una mediana de tiempo hasta respuesta tumoral de 4 meses, una mediana de la duración de la respuesta no alcanzada (superior a 26 meses, que fue la mediana de seguimiento) y una mediana de supervivencia libre de progresión de 22,1 meses; la tasa estimada de supervivencia a los 12 meses de tratamiento con sonidegib es del 100%. Aunque la supervivencia global no pudo estimarse por el alto porcentaje de pacientes censurados, parece que la calidad de vida percibida por los pacientes se mantiene estable durante el tratamiento y que el beneficio clínico perdura al menos hasta seguimientos de 42 meses.
En cuanto a su seguridad, el perfil toxicológico de sonidegib, aunque no es benigno, parece tolerable y clínicamente manejable –sobre todo, con ajustes posológicos–, en línea con el descrito para vismodegib (el otro inhibidor de Hh autorizado). Casi todos los pacientes (95%) sufrieron algún evento adverso, la mayoría leves y reversibles. Solo en el 23% de pacientes, los eventos adversos de grado 3/4 se relacionaron con el tratamiento. Las reacciones adversas más frecuentes fueron los espasmos musculares (49%, graves en 3,8%), alopecia (49%), disgeusia (44%), náuseas (39%), fatiga (33%), diarrea (32%), aumentos de CK (30%) y pérdida de peso (30%). El riesgo de carcinogenicidad por su uso a largo plazo no ha sido dilucidado completamente.
En definitiva, sin innovación en el plano mecanístico, parece altamente probable que sonidegib aporte una respuesta antitumoral de intensidad y duración clínicamente relevantes, sin empeorar la calidad de vida de los pacientes, pudiendo incluso aumentar las probabilidades de alcanzar una cirugía curativa. Puesto que no se dispone de comparaciones directas entre sonidegib y vismodegib, que las comparaciones indirectas sugieren que ambos fármacos pueden tener eficacias parejas y que la limitación de los resultados clínicos impide tener certeza sobre la magnitud y relevancia del beneficio, sonidegib no comporta una innovación disruptiva: representa una alternativa de tratamiento más en cuadros realmente infrecuentes pero muy complicados, con muy escasas opciones terapéuticas.
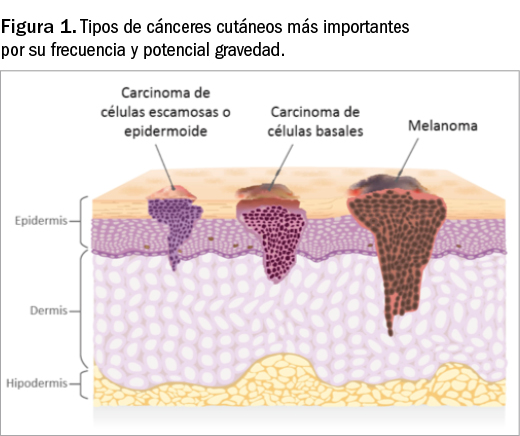
En global, el cáncer cutáneo representa la forma más frecuente de tumor maligno en la población caucásica (más de un tercio de todos los cánceres que afectan al ser humano)1. Con este término nos referimos todas las lesiones malignas que afectan a la piel y sus anejos; a grandes rasgos y considerando las diferencias epidemiológicas y pronósticas, se suelen dividir en dos grandes grupos: el melanoma y el cáncer cutáneo no melanoma (CCNM). En este último grupo se enmarcan las lesiones premalignas o carcinomas localizados in situ (queratosis actínica, enfermedad de Bowen, etc.) y las lesiones malignas más frecuentes en dermatología que derivan de los queratinocitos epidérmicos: el carcinoma basocelular y el carcinoma de células escamosas (Figura 1). Con una menor incidencia y características fisiopatológicas diferentes, así como tratamiento específico, se encontraría el carcinoma de células de Merkel.
El carcinoma de células basales o basocelular (CBC) –también denominado epitelioma basocelular o basalioma– es el subtipo de cáncer invasivo más común dentro de los CCNM, más incidente que el carcinoma de células escamosas o epidermoide (CE), y que representa el 75-80% de todos los casos de CCNM. De procedencia epitelial, a diferencia del melanoma (que deriva de los melanocitos), el CBC es la forma más común de cáncer cutáneo en los seres humanos (representa 8 de cada 10 casos), y se estima que el riesgo global de padecerlo a lo largo de la vida está entre un 28 y un 33%. Hay al menos 6 variantes histológicas principales de CBC, que incluyen: nodular (clásico), superficial, micronodular, morfeiforme (esclerosante), infiltrante y baso-escamoso, con diferencias de comportamiento (enfermedad agresiva vs. enfermedad menos agresiva).
Se califica como CBC localmente avanzado aquél en el que hay confirmación radiológica de invasión de determinadas estructuras vecinas en profundidad, y también probablemente aquél CBC de un tamaño e invasión suficientes (aunque no exista demostración radiológica de invasión en profundidad) en el que la cirugía y la radioterapia no son adecuadas, son insuficientes o están contraindicadas para lograr la curación del tumor, ya sea por características del propio tumor o del paciente. Suele caracterizarse por una larga evolución e historia de múltiples recurrencias.
El CBC ha experimentado un aumento persistente en las cifras de incidencia, que han adquirido proporciones epidémicas a pesar de los programas de educación sanitaria a nivel mundial. Algunos autores han estimado un incremento en la incidencia de todos los CCNM en torno a un 3-8% anual desde el año 1960. Sin embargo, debido a que estos tumores no suelen incluirse en registros oncológicos (como sí se incluye el melanoma), no se conoce la incidencia exacta del CBC, siendo muy probable que las cifras epidemiológicas –que suelen proceder de estudios realizados sobre poblaciones especialmente seleccionadas– estén subestimadas.
A modo de ejemplo, se estima que en los Estados Unidos se diagnostican al año más de 1,2 millones de casos de CBC, lo cual se traduce en tasas anuales de entre 407 y 485 casos por cada 100.000 habitantes en los hombres y de 212-253 casos/100.000 en las mujeres. En España, la tasa de incidencia bruta global anualizada ha sido estimada en 113,05 nuevos casos/100.000 habitantes para el CBC (y en 38,16 nuevos casos/100.000 habitantes para el CE), describiéndose, no obstante, notables diferencias de incidencia entre regiones; por ejemplo, el número de nuevos casos de CCNM en Madrid (175 casos/100.000) es superior al descrito en Granada (80 casos/100.000) (Tejera-Vaquerizo et al., 2016).
Por otra parte, el progresivo cambio demográfico con tendencias al envejecimiento en los países del “primer mundo” conlleva un aumento en las tasas de incidencia del CBC debido a que el riesgo de desarrollarlo se incrementa con la edad (probablemente como consecuencia directa de la acumulación de la exposición solar sobre la piel en el transcurso del tiempo). Una revisión sistemática de los estudios sobre la incidencia mundial de CBC (Lomas et al., 2012) demostró que las tasas de CBC han aumentado progresivamente en las últimas 4 décadas en un promedio de 20 casos nuevos más por cada 100.000 habitantes cada 15 años, es decir, un 5,5% de aumento por año, con grandes variaciones entre países.
Debido su capacidad de invasividad local, el CBC puede producir una morbilidad significativa, ya que la mayoría de los casos afectan a zonas funcionales visibles expuestas al sol y tienden a crecer lentamente. Aparece muy frecuentemente en la cara, pero puede aparecer en cualquier localización, siendo excepcional en palmas, plantas o mucosas. Sin embargo, afortunadamente, es muy infrecuente que se propague a otras partes del cuerpo (se detectan metástasis en menos del 0,6% de los casos, y mayoritariamente en nódulos linfáticos regionales y pulmones), aunque si no se trata, podría progresar a estadios avanzados, con invasión y destrucción de los tejidos próximos –más común en cara, cabeza y cuello, con la desfiguración física que ello supone– e incluso llegar a afectar a los huesos. Por tanto, a pesar de su menor incidencia, la mortalidad del CCNM es atribuible fundamentalmente al CE, el cual, aunque menos frecuente, comporta un riesgo mucho mayor de metástasis a distancia que el CBC.
Se considera que la exposición a los rayos ultravioleta (UV) –y, en mayor medida, a la radiación UV B– es el principal factor de riesgo de la mayoría de los cánceres de piel, incluyendo el CBC. En este sentido, la luz solar es la fuente principal de la radiación UV, aunque las lámparas utilizadas en los dispositivos (“camas”) bronceadores son otra fuente importante de rayos UV. El efecto negativo de estos reside en su capacidad para dañar la estructura del ADN de las células cutáneas, especialmente aquellas más superficiales, entre los que se incluyen los queratinocitos (y también los melanocitos).
La exposición a radiación ionizante o ciertos químicos (arsénico, brea de hulla, parafina, etc.), la piel sin color o hipocolorada (albinismo), la edad avanzada (es más frecuente en personas de entre 60 y 70 años), el sexo masculino (implica el doble de riesgo respecto al femenino), el cáncer de piel previo, la inflamación o lesión cutánea a largo plazo o grave (“quemaduras” pasadas), la xerodermia pigmentosa, la psoriasis y la inmunidad reducida2 (SIDA, tratamientos inmunosupresores, etc.) son reconocidos factores de riesgo en el desarrollo de CBC.
La lesión inicial del CBC suele manifestarse como una pequeña pápula indurada blancogrisácea recubierta por finas dilataciones de los capilares y vasos superficiales, conocidas como arañas vasculares o telangiectasias, que evoluciona a formas nodulares o, a veces, toma el aspecto de una cicatriz. Se puede manifestar como una úlcera que no cura y que muy lentamente va aumentando de tamaño (Figura 2); otras veces aparece como una lesión pigmentada semejante a un melanoma. Por su parte, el epitelioma basocelular superficial es una placa eritematosa que suele aparecer en tronco.
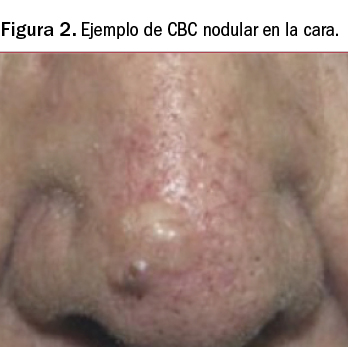
Como se ha sugerido, se trata de un tumor de crecimiento lento y solo de agresividad local (la aparición de metástasis es excepcional) pero, si se deja evolucionar sin tratamiento, es capaz de invadir estructuras subyacentes con importantes destrucciones de tejido, haciendo entonces muy difícil su tratamiento curativo. Cabe destacar que, incluso después del tratamiento, el CBC puede reaparecer en el mismo lugar de la piel; de hecho, en torno al 30-50% de las personas diagnosticadas con este tipo de tumor padecerán un nuevo cáncer de piel dentro de los siguientes 5 años. Por otro lado, las personas que han tenido CBC también tienen una probabilidad mayor de padecer nuevos cánceres en otros lugares de la piel.
El CBC guarda una estrecha relación con el tejido conectivo del entorno (se origina a partir de células madre indiferenciadas y pluripotentes de la capa basal epidérmica y folículos pilosebáceos), observándose en el mismo una mayor expresión del receptor para el factor de crecimiento derivado de plaquetas (PDGFR). Además, los fibroblastos podrían liberar citocinas que faciliten la supervivencia del epitelio tumoral; quizá esta vinculación explica la dificultad de aparición de metástasis.
Si bien lo más común es que este tipo de tumor aparezca de forma esporádica, en una proporción no desdeñable de casos juegan un rol clave los factores genéticos. Se han encontrado mutaciones en los genes supresores de tumores p53 y PTCH1, éste último asociado al síndrome del carcinoma basocelular nevoide (o síndrome de Gorlin3) que se hereda de forma autosómica dominante y se expresa con la aparición precoz de estos tumores, alteraciones en los huesos (quistes mandibulares) y pequeñas depresiones en palmas y plantas. La pérdida de función de PTCH1 resulta en un descontrol de la vía de señalización Hedgehog, que guarda una estrecha relación con el CBC.
El fenotipo Hedgehog (Hh) se descubrió en la mosca de la fruta (Drosophila malanogaster) como una mutación de polaridad de segmento, que proporciona información sobre la posición espacial durante el desarrollo embrionario de numerosos animales, incluyendo los seres humanos. En mamíferos existen tres genes homólogos de Hh que intervienen en una gran variedad de procesos embrionarios: Sonic (Shh), Indian (Ihh) y Desert (Dhh). El más estudiado es Shh que, además de jugar un papel importante en el desarrollo del sistema nervioso central (las mutaciones que inactivan Shh en seres humanos producen holoprosencefalia), está implicado en la formación de los folículos pilosos, donde residen las células madre de la epidermis.
En la vía de señalización Hedgehog intervienen cuatro proteínas importantes: Patched (ptch), Smoothened (smo), Hedgehof (hh) y gli. El gen PTCH codifica una proteína presente en la membrana celular (con 12 sectores de transmembrana), que se asocia con la proteína smo, también presente en la superficie celular (con 7 sectores de transmembrana), formando un complejo que funciona como un receptor para el factor de crecimiento hh. En ausencia de hh (en la fase G0 del ciclo celular), ptch y smo forman un complejo inactivo mediante el cual ptch inhibe a smo, que es la molécula que tiene actividad en la señalización intracelular. Por contra, cuando hh se une a ptch, smo se libera del freno impuesto por ptch y envía una señal al núcleo de la célula que conduce a la activación/represión de ciertos genes.
Las mutaciones en ptch activan constitutivamente la señalización de la vía Hedgehog y conducen a alteraciones moleculares relacionadas con el cáncer. El factor de transcripción gli, activado por esta vía de señalización, promueve la proliferación celular. En condiciones normales, este factor se encuentra secuestrado en un complejo citoplasmático formado por proteínas asociadas al citoesqueleto como suf o fused. Cuando la vía se activa, gli se libera del complejo y en el núcleo regula la transcripción de sus genes diana (Wnt, BMPs, PDGFR, FOXE1, etc.). Gli está sobreexpresada en todos los tumores en los que está implicada la vía de señalización Hedgehog y, de hecho, las mutaciones en ptch son muy frecuentes (80-90%) en los CBC esporádicos. También se han encontrado con cierta frecuencia mutaciones de smo en estos tumores (10%), actuando como un oncogén. En definitiva, se espera que la mayoría de casos de CBC dependan de esta vía de señalización para su crecimiento y progresión.
El CBC es normalmente susceptible de tratamiento local, cuyo objetivo principal es la curación del proceso, que se consigue en la mayor parte de los casos. En general, la primera línea de tratamiento, en cualquier cáncer cutáneo –y del CBC en particular– es la cirugía del cáncer in situ con la obtención de márgenes libres de tumor. Alternativamente, se puede utilizar radioterapia u otros tratamientos médicos locales. La terapia sistémica, como norma general, se puede utilizar cuando no son tributarios de cirugía y/o radioterapia y son suficientemente invasivos o están diseminados.
En cuanto a la cirugía, se dispone de técnicas de cirugía convencional (extirpación simple por sutura directa o técnicas de injertos) y de cirugía con control de márgenes, como la cirugía micrográfica de Mohs. Esta última técnica es el método de referencia en el tratamiento del CBC en la actualidad, pues consigue las mejores tasas de curación (cercanas al 90% a los 5 años) y resultados en términos de recurrencia tumoral, especialmente en aquellos pacientes en los que, debido a la localización del tumor (habitualmente en la cara), se precisa preservar la mayor cantidad de tejido sano alrededor de la lesión. Se basa en la combinación de una evaluación microscópica de los márgenes del tumor con cartografía detallada de la orientación del tumor. Resulta especialmente útil en el tratamiento de tumores grandes, así como en aquellos en los que los bordes no están bien definidos, tumores localizados en o cerca de la nariz, los ojos, las orejas, la frente, el cuero cabelludo, los dedos y el área genital, así como aquellos que han reaparecido después de otros tratamientos.
La criocirugía (con nitrógeno líquido) puede usarse en algunos CBC pequeños, pero usualmente no se recomienda para tumores más grandes, ni para tumores localizados en ciertas partes de la nariz, las orejas, los párpados, el cuero cabelludo o las piernas. A menudo, la radioterapia es una buena opción en el tratamiento de pacientes que no pueden tolerar la cirugía y para tumores que comprometen los párpados, la nariz o las áreas de las orejas que pueden ser difíciles de tratar quirúrgicamente. El curetaje, el láser y la electrodesecación son otros tratamientos comunes para los CBC pequeños, aunque puede ser necesario repetirlos para asegurar el éxito completo.
La probabilidad de que el CBC recurra varía desde menos de 5% para la cirugía Mohs hasta 15% o más para algunos otros tratamientos, pero esto depende del tamaño del tumor. Los tumores pequeños tienen menos probabilidad de recurrir que los tumores más grandes. Incluso si el tumor regresa, a menudo se puede retratar eficazmente. En una revisión sistemática de 27 ensayos controlados aleatorios que comparaban diferentes tratamientos (Bath-Hextall et al., 2007) se indicó que solo el 50% de las recidivas se presentan dentro de los primeros 2 años, el 66% después de 3 años y 18% después de 5 años. En general, se considera que las tasas de recurrencia a 10 años son casi el doble de las tasas de recurrencia a 2 años.
El tratamiento farmacológico del CBC suele emplearse en tumores de gran tamaño y localizado en áreas fotoexpuestas, para sus formas superficial y nodular, evitándose en aquellos tipos histológicos de comportamiento biológico más agresivo como el esclerodermiforme o morfeiforme, o el tipo histológico infiltrativo; en estos últimos resulta más conveniente y adecuada la resección quirúrgica para evitar las recidivas por persistencia de células tumorales viables. El objetivo, en todo caso, debería ser eliminar el tumor sin que haya secuelas funcionales ni estéticas, alcanzando la curación.
En la elección de la farmacoterapia deberá tenerse en cuenta el tipo y la localización del tumor, así como las características del paciente y los factores de riesgo de recurrencias, aunque la evidencia de estudios controlados y aleatorizados con seguimiento a 5 años que comparen la eficacia de los diferentes tratamientos es muy escasa. Entre las distintas opciones terapéuticas autorizadas con indicación de carcinoma basocelular, se dispone de:
Sonidegib se une específicamente a smo de forma que inhibe la activación y liberación de los factores Gli mediadas por ésta. En consecuencia, bloquea la vía de señalización y, con ello, la expresión de genes implicados en el crecimiento del tumor. En ensayos de competición de la unión in vitro, sonidegib ha demostrado la capacidad de desplazar a ligandos la proteína smo con una IC50 de 11 nM y ha demostrado también carecer de afinidad significativa de unión a un gran número de dianas evaluadas, entre ellas otros receptores acoplados a proteínas G, canales iónicos, receptores y transportadores nucleares.
Cabe destacar que en los ensayos in vivo y clínicos, la respuesta tumoral a sonidegib fue independiente de su dosis y de su concentración plasmática (en el intervalo de dosis de 200-800 mg). En tejidos tumorales frescos de pacientes con CBC localmente avanzado tratados con el fármaco, se demostró que la inhibición media de Gli-1 se situaba entre el 82% y el 92% con la dosis de 200 mg/día y mayor del 95% a la dosis de 800 mg/día; pero tampoco se encontró una correlación entre esos niveles de inhibición con la respuesta al tratamiento.
Por último, aunque no se dispone de datos en mujeres embarazadas, los estudios realizados en animales apuntan a que sonidegib tiene un elevado potencial teratogénico, que sería –como en el caso de vismodegib– coherente con su mecanismo de acción, ya que el fenotipo Hedgehog (hh) proporciona información de posición espacial durante el desarrollo embrionario de numerosos animales, incluyendo los seres humanos. Debido a ello, el fármaco puede provocar una notable toxicidad fetal a lo largo de todo el embarazo e incluso afectar al desarrollo posnatal. En este sentido, con exposiciones próximas a las previstas en humanos, en animales de experimentación (ratas, perros y conejos) se han descrito defectos irreversibles sobre el crecimiento de los dientes, cierre prematuro de la epífisis femoral, efectos sobre el aparato reproductor masculino y femenino (incluyendo abortos y resorción fetal), atrofia de los folículos pilosos (alopecia), pérdida de peso corporal y efectos sobre los nódulos linfáticos. Sonidegib está contraindicado en embarazo.
El sonidegib es una pequeña molécula sintética, de carácter policíclico, que guarda cierta similitud estructural –aunque menor de la que cabría esperarse por su coincidencia de mecanismo de acción– con vismodegib (Figura 3). Este último fármaco, que emergió como cabeza de serie de este grupo de productos con potencial antineoplásico, deriva de las observaciones realizadas en la década de los 50 del pasado siglo realizadas con la ciclopamina5, un alcaloide esteroídico presente en la planta Veratrum californicum, que está estructuralmente relacionado con la jervina (de hecho, vismodegib es la 11-desoxojervina).
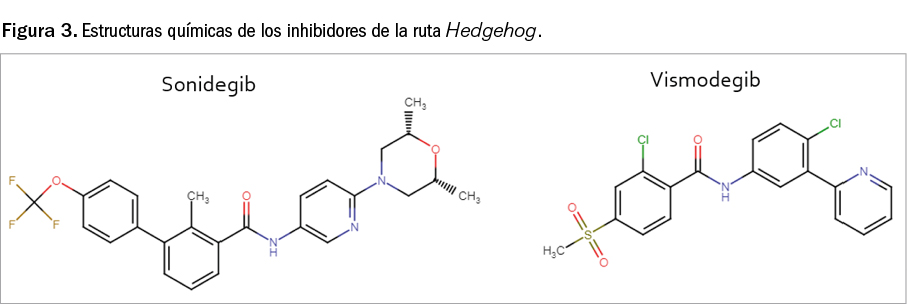
El nombre químico de sonidegib es N-[6-(cis-2,6-dimetilmorfolin-4-il)piridina-3-il]-2-metil-4’-(trifluorometoxi)-[1,1’-difenil]-3-carboxamida, y se corresponde con una fórmula química de C26H26F3N3O3 y un peso molecular de 485,5 g/mol. Se trata de una molécula no quiral en la que se han identificado múltiples polimorfos, de los cuales el empleado en el desarrollo clínico del medicamento es un co-cristal de monofosfato de sonidegib con ácido fosfórico. En su forma de difosfato se presenta en forma de polvo no higroscópico, cristalino, de color blanco a amarillo claro, prácticamente insoluble en medio acuoso a pH entre 1 y 7,5, ligeramente soluble en acetona y poco soluble en alcoholes.
La eficacia y seguridad clínicas de sonidegib por vía oral han sido evaluadas en la indicación y dosis autorizadas mediante un ensayo clínico pivotal de fase II (exploratorio de eficacia y seguridad) con un diseño aleatorizado (2:1), doblemente ciego, multicéntrico y multinacional (58 centros en 12 países) y comparativo de dos niveles de dosis del fármaco (ensayo BOLT / A2201).
Dicho estudio incluyó un total de 230 pacientes adultos (≥18 años) que presentaban bien carcinoma basocelular localmente avanzado (CBCla, N= 194) o bien carcinoma basocelular metastásico (CBCm, N= 36) y que no eran candidatos a recibir radioterapia, cirugía u otros tratamientos locales; de ambos grupos, 15 y 1 pacientes presentaban diagnóstico de síndrome de Gorlin. Entre los criterios de inclusión destaca que los pacientes debían presentar un buen estado funcional (puntuación ECOG6 ≤2) y una adecuada funcionalidad renal, hepática y de la médula ósea. Por el contrario, se excluyeron pacientes que habían sido sometidos a cirugía en las 4 semanas previas al inicio del estudio, que no podían tomar medicación por vía oral, que hubieran recibido tratamiento previo con inhibidores de la vía Hh o hubieran estado bajo cualquier tratamiento antineoplásico en las 4 semanas anteriores, pacientes con trastornos de tipo neuromuscular o mujeres embarazadas/lactantes.
Los pacientes fueron aleatorizados (2:1) a recibir sonidegib 800 mg/día (N= 151) o 200 mg/día (N= 79). En el brazo de 800 mg se permitieron 2 reducciones de dosis pero en el brazo de 200 mg solo se permitió un ajuste posológico, suspendiendo el tratamiento si se necesitaba una segunda reducción. Las características basales del conjunto de los pacientes estuvieron bastante balanceadas entre los dos brazos del estudio. La mediana de edad fue de 66 años (rango 24-93, con el 54,3% de ≥65 años), 63% eran varones, el 94% de raza blanca (el 56% eran europeos y el 39% norteamericanos), y la mayoría de los pacientes tenían un estado funcional ECOG de 0 (63%) o 1 (27%). En el brazo de sonidegib 200 mg/día, el 84% (66) de los pacientes presentaban CBCla (47% con histología agresiva y 37% con histología no agresiva) y el 16% (13) presentaba CBCm. Destaca también que un 80% de los pacientes con CBCla había recibido tratamientos previos (de ellos, el 79% cirugía, 8% radioterapia y 5% tratamientos antineoplásicos).
La variable principal de eficacia fue la tasa de respuesta objetiva (TRO) según criterios RECIST modificados en pacientes con CBCla y según criterios RECIST 1.17 en pacientes con CBCm, según un revisor central. Siguiendo dichos criterios, como variables secundarias de eficacia se determinaron la duración de la respuesta (DR), el tiempo hasta la respuesta tumoral (TRT) y la supervivencia libre de progresión (SLP); también se estimaron todas esas variables y la supervivencia global (SG) según evaluación por el investigador local. El tratamiento con sonidegib se mantuvo hasta progresión de la enfermedad o toxicidad inaceptable.
El análisis primario de los datos clínicos se llevó a cabo a los 12 meses (con una mediana de seguimiento de 13,9 meses) en la población total del estudio (conjunto de análisis completo o FAS, full analysis set, que incluye a todos los pacientes aleatorizados o población por intención de tratar) y en la población evaluable de eficacia (conjunto de análisis de eficacia primario o pEAS, que incluye todos los pacientes con CBCla evaluados adecuadamente por resonancia magnética o fotografías digitales y todos los pacientes con CBCm).
No obstante, la siguiente tabla (Tabla 1) refleja los principales resultados de eficacia en la población FAS con un seguimiento a 18 meses (la última actualización de resultados considerada en la autorización del medicamento), para el brazo de dosis de 200 mg/día de sonidegib, la que fue finalmente autorizada (descartándose la dosis de 800 mg/día por sus peor perfil beneficio-riesgo), y para población de pacientes con CBCla, la única incluida en la indicación autorizada (no se verificó una eficacia significativa en la población de pacientes con CBCm) (EMA, 2015). En ese punto temporal, 11 pacientes (14%) del brazo de sonidegib 200 mg continuaban en tratamiento, 1 paciente (1,3%) había fallecido, 27 (34%) habían progresado, y 22 (28%) habían interrumpido el tratamiento por eventos adversos.
De forma exploratoria, se evaluó la influencia del tratamiento sobre la calidad de vida percibida por los pacientes. Empleando el módulo específico para cáncer de cabeza y cuello del Cuestionario de Calidad de Vida Core 30 de la Organización Europea para la Investigación y el Tratamiento del Cáncer (EORTC QLQ-C30, H&N35), se observó que la mayoría de pacientes reportaban un mantenimiento y/o mejoría de los síntomas relacionados con la enfermedad, la funcionalidad y el estado de salud con sonidegib, aunque aquellos tratados con la dosis de 800 mg notificaron mayor fatiga, dolor y pérdida de peso.
Se debe mencionar que, adicionalmente, también se han desarrollado dos estudios clínicos en población pediátrica (hasta 62 pacientes) con otros tipos de tumores: i) el ensayo CLDE225X2104 de fase I/II con pacientes con meduloblastoma recurrente o refractario u otras neoplasias malignas probablemente dependientes de la vía de señalización Hh; y ii) el estudio CLDE225C2301 de fase II, multicéntrico, abierto y de grupo único, con pacientes con meduloblastoma recidivante activado por Hh. No obstante, no se ha podido confirmar la eficacia de sonidegib en esta población, incluso a pesar de la estrategia de enriquecimiento en meduloblastoma activado por Hh.
Por otro lado, la seguridad clínica del nuevo fármaco ha sido caracterizada en base a datos de 293 pacientes tratados durante todo su desarrollo clínico, incluyendo los 230 del ensayo pivotal. En este último, con una duración media del tratamiento de 11 meses con la dosis autorizada de 200 mg, el 95% de pacientes sufrió algún evento adverso, si bien solo en el 30,4% fueron graves (grado ≥3) y en solo el 22,8% estos eventos graves se relacionaron con el tratamiento. Sin embargo, el 36,1% de pacientes requirió la interrupción y/o reducción de dosis por eventos adversos (el 21,5% interrumpió el tratamiento).
Las reacciones adversas más frecuentemente notificadas –de cualquier grado– con sonidegib fueron: espasmos musculares (49,4%), alopecia (43%), náuseas (32,9%), disgeusia (38%), fatiga (29,1%), dolor musculoesquelético (29,1%), diarrea (24,1%), aumento de la creatinina cinasa (CK) (29,1%), pérdida de peso (26,6%), falta de apetito (19%), mialgia (19%), dolor abdominal (15,2%) y cefalea (15,2%). En general, fueron de carácter leve-moderado, destacando por su gravedad (grado 3/4) las elevaciones de la CK (6,3%) y de la lipasa (5%), los espasmos musculares (2,5%) y otras más inespecíficas, como artralgia, alopecia, náuseas, estreñimiento, vómitos y disminución de peso (1,3% cada una, notificadas en un único paciente). La mayoría pudieron manejarse adecuadamente con ajustes de dosis, o con tratamientos farmacológicos y no farmacológicos. Los eventos adversos que supusieron suspensión del tratamiento de sonidegib 200 mg/día con mayor frecuencia fueron espasmos musculares (3,8%), disgeusia (2,5%), pérdida de peso (2,5%) y náuseas (2,5%). Los pacientes en tratamiento con sonidegib 800 mg/día interrumpieron más precozmente el tratamiento por eventos adversos, que en general tuvieron mayor incidencia y gravedad.
Por último, hay que destacar que en el análisis a 18 meses, 1 paciente del brazo con sonidegib 200 mg había fallecido (1,3%) y 3 (3,8%) habían desarrollado carcinoma cutáneo de células escamosas (CE). Se notificaron también casos de melanoma maligno, cáncer de próstata, linfoma de células B y cáncer de vulva, si bien no se ha establecido una relación directa de sonidegib con estos eventos. También hay que recordar, dentro del perfil toxicológico del nuevo fármaco, su potencial teratógeno y de fetotoxicidad, que hacen necesario el uso de métodos anticonceptivos eficaces durante el tratamiento y hasta 6 meses después (pues sonidegib tienen una vida media de 28 días).
Los resultados de eficacia y de seguridad actualizados tras seguimientos de 30 meses (Lear et al., 2018) y a 42 meses (Dummer et al., 2019) están en línea y son prácticamente idénticos a los comentados tras los análisis iniciales.
El sonidegib es un nuevo inhibidor de la ruta Hedgehog (Hh), capaz de unirse a la proteína transmembrana Smoothened (smo) e impedir la activación y localización nuclear de los factores de transcripción gli; así, bloquea la vía de señalización y la expresión de los genes diana Hh e inhibe la proliferación, diferenciación y supervivencia de las células tumorales mediadas por dicha ruta en hasta el 90% de los tumores cutáneos de células basales. En base a ese mecanismo, el medicamento ha sido oficialmente autorizado para el tratamiento por vía oral de pacientes adultos con carcinoma basocelular localmente avanzado (CBCla) que no es susceptible a la cirugía curativa ni a radioterapia.
Los datos de eficacia y seguridad clínicas conducentes a su autorización son limitados y metodológicamente no demasiado robustos. Derivan en su práctica totalidad de un único ensayo clínico de fase 2, aleatorizado, abierto y doblemente ciego, que ha incluido un total de 230 pacientes adultos con CBC localmente avanzado (N= 194) o metastásico (N= 36) y ha evaluado 2 dosis del fármaco (200 mg/día y 800 mg/día). No obstante, la autorización final no ha incluido los tumores metastásicos por ausencia de eficacia significativa y también ha excluido la dosis de 800 mg/día por una mayor toxicidad. La EMA considera aceptable el diseño de este estudio pivotal (representatividad de la muestra de pacientes y selección de las variables de eficacia), si bien la ausencia de control –placebo o comparador activo– supone la mayor limitación en la interpretación de los datos.
En cualquier caso, se asume que esas limitaciones se superan por la significación de los datos de eficacia. Tras el análisis de los resultados a los 18 meses, la tasa de respuesta objetiva (TRO) en el conjunto de pacientes con CBCla aleatorizados a recibir sonidegib 200 mg/día se situó en el 56,1% (4,6% de respuestas completas y 51,5% de respuestas parciales) en la revisión por un comité central independiente (variable principal), siendo incluso mayor en la revisión por el investigador (TRO= 71,2%). Las variables secundarias respaldan, pues, la eficacia de sonidegib, con una mediana de tiempo hasta la respuesta tumoral de 4 meses, una mediana de la duración de la respuesta no alcanzada (y, por tanto, superior a 26 meses, que fue la mediana de seguimiento) y una mediana de supervivencia libre de progresión de 22,1 meses; todas ellas según revisión central independiente. La tasa estimada de supervivencia a los 12 meses de tratamiento con sonidegib es del 100% para los pacientes con CBCla.
Aunque la supervivencia global no pudo estimarse por el alto porcentaje de pacientes censurados, destaca el hecho de que la mayoría de pacientes refieren un mantenimiento/mejoría de la calidad de vida durante el tratamiento, y que el beneficio clínico con el nuevo fármaco se mantiene al menos hasta seguimientos de 42 meses. La eficacia del nuevo fármaco parece ser independiente del estado de progresión de la enfermedad en la aleatorización, pero no se ha realizado un estudio exhaustivo por subgrupos. Las notables diferencias encontradas entre las variables dependiendo del comité evaluador (central independiente vs. investigadores del estudio), por ejemplo, en las tasas de respuesta objetiva (56,1% vs. 71,2%) tampoco contribuyen a eliminar las incertidumbres y establecer el verdadero valor terapéutico de sonidegib en su indicación.
Por otra parte, a la dosis autorizada y en base al limitado número de pacientes tratados, sonidegib parece un fármaco bien tolerado: presenta una toxicidad a tener en cuenta, pero que puede manejarse clínicamente con ajustes posológicos (necesarios en casi 1 de cada 3 pacientes tratados) o tratamientos específicos. En el ensayo pivotal, casi todos los pacientes (95%) sufrieron algún evento adverso, que fueron graves en aproximadamente el 30%; en el 23% de pacientes, estos eventos de grado 3/4 se relacionaron con el tratamiento. Los EA más frecuentes –y, en su mayoría, reversibles– con sonidegib 200 mg fueron espasmos musculares (49%, graves en 3,8%), alopecia (49%), disgeusia (44%), náuseas (39%), fatiga (33%), diarrea (32%), aumentos de CK (30%) y pérdida de peso (30%). Además, el riesgo de carcinogenicidad por su uso a largo plazo no ha sido dilucidado completamente.
El perfil toxicológico es, pues, consistente con el de vismodegib (el otro inhibidor de Hh), siendo la toxicidad muscular –evidenciada por espasmos musculares, rabdomiolisis o aumento de los niveles sanguíneos de CK– la reacción adversa más relevante, la cual puede considerarse como un efecto de clase8. También hay que recordar, en este punto, el alto potencial teratógeno de sonidegib y su contraindicación en embarazo y lactancia, así como su riesgo de interacción con inhibidores o inductores potentes de CYP3A4, por ser ésta su principal vía de metabolización.
No se dispone de comparaciones directas entre la eficacia y seguridad de sonidegib y vismodegib. Las comparaciones indirectas, con limitaciones intrínsecas para la extracción de conclusiones, presentan la problemática adicional de que en ambos casos los estudios han sido no comparativos y sin brazo control. A pesar de ello, algunos trabajos publicados sugieren que sonidegib parece comparable a vismodegib en términos de actividad antitumoral en pacientes con CBCla, con una eficacia clínicamente relevante en términos de TRO: se ha sugerido que las TRO ajustadas alcanzan de media el 56,7% con sonidegib vs. 47,6% con vismodegib; la mediana de SLP sí se estima mayor en el caso de sonidegib (22,1 vs. 9,5 meses) (Odom et al., 2017).
Por último, recientes evidencias sugieren que, a pesar de la buena respuesta inicial, los pacientes tratados con inhibido res de la vía Hh han desarrollado con el tiempo resistencias por el desarrollo de mecanismos de compensación (tasas de nuevos crecimientos tumorales >20%). No se conoce aún la eficacia de administrar sonidegib en pacientes que han fracasado al tratamiento con vismodegib, ya que la resistencia a uno podría conferir resistencia al otro. Una evaluación individualizada del beneficio-riesgo permitirá esclarecer estas incertidumbres, determinando la conveniencia del tratamiento con este tipo de fármacos en pacientes recidivantes (AEMPS, 2019).
En resumen, sonidegib es un nuevo inhibidor de smo y la ruta de señalización que, sin suponer ninguna novedad mecanística, aporta un beneficio cosmético y funcional significativo en pacientes con CBC avanzado no susceptible de cirugía curativa o radioterapia, esto es, cuadros realmente infrecuentes pero muy complicados y con muy escasas opciones terapéuticas. A pesar de que hay incertidumbres sobre el perfil beneficio-riesgo a largo plazo, parece altamente probable que sonidegib aporte una respuesta antitumoral de intensidad y duración clínicamente relevantes, sin empeorar la calidad de vida de los pacientes, pudiendo incluso aumentar las probabilidades de realizar una cirugía curativa. La limitación de los resultados clínicos (fase 2 y sin comparador) impide tener certeza sobre la magnitud y relevancia del beneficio, así como posicionar a sonidegib respecto a vismodegib, quedando ambos como alternativas terapéuticas similares.