CÓMO LOCALIZAR CAMBIOS DE NOMBRE Y DE LABORATORIO CON BOT PLUS
Además de la información que se incluye en los listados mensuales publicados en PAM, en BOT PLUS se incluye un apartado de Histórico, en las fichas de medicamentos, en el que se presenta información referente a cambios que haya sufrido anteriormente el medicamento o producto, entre otros, los cambios de nombre y los cambios de laboratorio. Esta información también está disponible para productos sanitarios financiados o dietoterápicos.
Se añade la posibilidad de visualización de las situaciones anteriores (o incluso futuras) relacionadas con un cambio de nombre.
Con automatismos que nos permiten localizar un medicamento que haya cambiado de nombre, independientemente de cuál usemos.
Posibilidad de generar listados por Histórico
Además de la información existente en Histórico, se permite la explotación de la información incluida en BOT PLUS en este apartado, mediante la integración de la información almacenada en Histórico en el apartado de Listados de BOT PLUS, que permite realizar consultas entre rangos de fechas y por un concepto en concreto de entre los almacenados en el apartado de Histórico. Entre ellos se incluyen, precisamente, los conceptos “Cambio del nombre del medicamento” y “Cambio del laboratorio comercializador”.
Es importante indicar que se valora el grado de innovación. Todos los medicamentos, sean innovadores o no, tienen utilidad terapéutica, en tanto que su autorización por las autoridades sanitarias implica que han demostrado rigurosamente su eficacia, su seguridad, su calidad y las condiciones de uso (incluyendo la información contenida en la ficha técnica – sumario de características – y en el prospecto del medicamento). Por tanto, la valoración que se hace se refiere a la incorporación, en el grado que se determine, de algún elemento innovador con respecto a otros medicamentos autorizados previamente para iguales o similares indicaciones terapéuticas o, en su caso, cubriendo la ausencia de éstas.
Asimismo, debe considerarse que ésta es una evaluación que se practica coincidiendo con la comercialización inicial del medicamento. Se trata, por consiguiente, de una valoración provisional de la innovación realizada en función de la evidencia clínica disponible hasta el momento, lo que no prejuzga, en ningún caso, la disponibilidad posterior de nuevas evidencias científicas (de eficacia o de seguridad) en la indicación autorizada o el potencial desarrollo y autorización, en su caso, de nuevas indicaciones terapéuticas o la imposición de restricciones de uso en las anteriores.
Se consideran tres posibles niveles, adjudicados en función de la relevancia de la(s) innovación(es) presentes en el nuevo medicamento, siempre en relación al arsenal terapéutico disponible clínicamente en España en el momento de la comercialización:
SIN INNOVACIÓN (*). No implica aparentemente ninguna mejora farmacológica ni clínica en el tratamiento de las indicaciones autorizadas.
INNOVACIÓN MODERADA (**). Aporta algunas mejoras, pero no implica cambios sustanciales en la terapéutica estándar.
INNOVACIÓN IMPORTANTE (***). Aportación sustancial a la terapéutica estándar.
Se distinguen dos niveles de evidencia científica para los aspectos innovadores de los nuevos medicamentos:
Evidencia clínica: mediante estudios controlados, específicamente diseñados y desarrollados para demostrar la eficacia y la seguridad del nuevo medicamento, con demostración fehaciente de lo que puede ser un avance o mejora sobre la terapia estándar hasta ese momento, en el que caso de que exista.
Plausibilidad científica (potencialidad): existencia de aspectos en el medicamento que teórica y racionalmente podrían mejorar la terapéutica actual, pero que no han sido adecuadamente demostrados mediante ensayos clínicos, bien por motivos éticos o bien por imposibilidad de realización en el momento de la comercialización del nuevo medicamento: perfil de interacciones, mecanismos nuevos que permiten nuevas vías terapéuticas, nuevos perfiles bioquímicos frente a mecanismos de resistencia microbiana, posibilidad de combinar con otros medicamentos para la misma indicación terapéutica, efectos sobre el cumplimiento terapéutico (por mejoras en la vía, número de administraciones diarias, etc.), mejora de la eficiencia económica, etc.
El rigor de los datos contrastados mediante ensayos clínicos controlados (evidencia clínica) es determinante en la valoración de la innovación, mientras que las potencialidades solo pueden ser valoradas accesoriamente, como aspectos complementarios de esta valoración. En ningún caso, un medicamento es valorado con un nivel de innovación importante en función de sus ventajas potenciales, si no aporta otras ventajas demostradas clínicamente. Se analizan cinco aspectos de la innovación: clínica, molecular, toxicológica, físico-química y económico-tecnológica. Como ya se ha indicado, la fundamental y determinante es la novedad clínica.
El trifaroteno es un nuevo derivado de retinoide de cuarta generación que actúa como un potente agonista del receptor γ del ácido retinoico (RARγ), con una especificidad muy elevada y una afinidad 50 veces mayor que sobre RARα y 8 veces mayor que sobre RARβ; no presenta, en cambio, actividad sobre receptores X de retinoides (RXR). Así, puesto que los RARγ son los receptores de retinoides más abundantes en la piel, trifaroteno será capaz de modular la expresión de genes diana implicados en el crecimiento y la diferenciación celular, la respuesta al estrés, cascadas antiinflamatorias y la apoptosis, entre otros procesos. Si bien el mecanismo exacto por el cual mejora el acné no se conoce completamente, el medicamento ha sido autorizado para el tratamiento (por vía tópica en forma de crema) de acné vulgar en la cara, en el pecho y/o en la espalda en pacientes mayores de 12 años, en presencia de numerosos comedones, pápulas y pústulas.
Su eficacia y seguridad clínica han sido bien definidas en 2 ensayos pivotales de fase 3 de igual diseño, doble ciegos, de grupos paralelos y controlados por vehículo, que aleatorizaron un total de 2.420 pacientes de > 9 años de edad con acné facial y troncal moderado (grado 3). Con una aplicación tópica diaria durante 12 semanas, el fármaco demostró una superioridad significativa frente a la crema vehículo en pacientes de ≥ 12 años, evidenciada por tasas de éxito terapéutico del 29-42% en acné facial (vs. 19-26% con vehículo) y del 36-43% en acné troncal (vs. 25-30%). Además, la reducción de las lesiones inflamatorias (en un 54-66%) y no inflamatorias (en un 49-58%) en ambas localizaciones también fue significativamente superior al vehículo; el beneficio clínico era evidente incluso desde la primera-segunda semana de tratamiento. Un estudio adicional de un único brazo con datos de tratamiento durante 1 año en 342 pacientes confirmó los resultados de los estudios pivotales y el beneficio continuo del tratamiento con trifaroteno: las tasas de éxito aumentaban hasta el 65% para el acné en cara (desde el 27% a la semana 12) y hasta el 67% para el acné en tronco (desde el 39%), con una amplia proporción de pacientes (54%) que reportaban que el acné no afectaba a su calidad de vida.
Con respecto a la seguridad, su perfil toxicológico está en línea con el ya conocido para otros retinoides tópicos. Ha sido bien caracterizado y parece aceptable (tasas de discontinuación bajas, de < 2% a las 12 semanas) y clínicamente manejable con ajustes posológicos o el uso de hidratantes/limpiadores. El tratamiento con la crema de trifaroteno se asocia con la aparición signos y síntomas locales leves-moderados (eritema, descamación, sequedad y ardor/quemazón), de incidencia variable (10-30%) y transitorios; la tolerabilidad fue mejor en tronco que en cara. Las reacciones adversas más frecuentes son: irritación (7,5% vs. 0,3% con vehículo) y prurito en el lugar de aplicación (2,4% vs. 0,8%) y quemaduras solares (2,6% vs. 0,5%). Su escasa absorción sistémica y rápido metabolismo hepático hacen que carezca de eventos adversos sistémicos, permitiendo su uso en amplias áreas cutáneas del tronco. Pero se mantiene la contraindicación de su uso en embarazo por el riesgo de teratogenicidad.
En ausencia de comparaciones directas con otras alternativas de tratamiento tópico del acné en adolescentes y adultos y, en particular, frente a los otros retinoides disponibles desde hace décadas (tretinoína y adapaleno), resulta difícil su posicionamiento. Parece que trifaroteno podría situarse como una alternativa más en primera línea del tratamiento tópico del acné moderado, aportando un beneficio clínico similar o ligeramente superior a otros fármacos disponibles. Aunque no representa una innovación importante en términos mecanísticos, aporta como novedad respecto al resto de retinoides su selectividad por el receptor RARγ cuya significación clínica no está del todo clara; su reducida afinidad sobre los RARβ dérmicos podría relacionarse con una menor incidencia de irritación cutánea, lo cual aún debe ser comprobado. Conviene subrayar que es la primera incorporación en las últimas dos décadas al grupo bien conocido de los retinoides y el primer fármaco que ha demostrado eficacia significativamente superior a placebo –en estudios bien diseñados– en acné del tronco, que hasta ahora representaba una necesidad médica no cubierta. No representa una cura del acné pero permite un buen control de los síntomas con una tolerabilidad aceptable.
Aspectos fisiopatológicos
El acné es una enfermedad inflamatoria (dermatosis) de la piel de la cara y de la parte superior del tronco. Se caracteriza por la aparición de seborrea, comedones y lesiones inflamatorias tanto superficiales, pápulas y pústulas, como más profundas, nódulos y quistes, pudiendo dejar lesiones residuales como manchas y cicatrices. Se manifiesta en áreas de la piel ricas en folículos pilosebáceos, afectando sobre todo a la cara y, en menor grado, a los hombros, la parte superior del tórax y la espalda, principalmente en adolescentes.
Suele presentarse como un trastorno dermatológico leve, que generalmente no deja secuelas y que se controla o desaparece de manera espontánea. No obstante, la posibilidad real de desarrollar lesiones crónicas, inflamatorias e incluso cicatriciales en ciertas formas crónicas y agresivas, puede producir la aparición de trastornos psicológicos, de relación y de autoestima importantes.
Básicamente, se distinguen dos tipos de lesiones en el acné: inflamatorias y no inflamatorias. Entre las primeras, las espinillas, comedones cerrados o microquistes constituyen el elemento más característico (Figura 1), y lo que define al acné. Provienen del aumento del sustrato córneo del conducto pilosebáceo, lo que da lugar a una obstrucción del mismo. Se trata de una lesión levemente elevada, coronada por un punto negro de consistencia córnea, que al ser comprimido libera una masa pastosa y blanquecina, que es el sebo albergado en el folículo. Por presión de la masa retenida sobre la pared folicular, se produce la ruptura de la misma y la consiguiente invasión del material hacia la dermis, iniciándose el proceso inflamatorio, que se traduce en la aparición de lesiones típicamente inflamatorias: pápulas1, pústulas2, nódulos, quistes (redondeados, formados a base de queratina) y, posteriormente, cicatrices. Estas últimas no siempre van ligadas a una manipulación de las lesiones ni tampoco a la gravedad del acné.
En cambio, los comedones abiertos no se encuentran siempre ni acostumbran a presentar cambios inflamatorios (salvo que se manipulen de forma inadecuada). Su aspecto es debido a la compactación de células foliculares en el ducto y a la oxidación del sebo y la melanina, no a la suciedad.
Los nódulos se encuentran en un plano más profundo de la piel y pueden ser fluctuantes cuando se palpan (son más palpables que visibles). Son lesiones infiltrativas que representan la inflamación de todo el folículo y la dermis circundante, recubierta por piel normal, que evoluciona lentamente hacia la inflamación y la resolución, responsable de la mayoría de las cicatrices (que pueden ser de todo tipo: atróficas, irregulares o queloideas) e hiperpigmentación.
La presencia de unas u otras lesionesdepende de la profundidad y de la intensidad de la reacción inflamatoria. Cuando ésta se produce con profundidad, el resultado es la aparición de pápulas profundas, nódulos o quistes, e incluso fístulas. En la evolución normal del proceso pueden aparecer máculas pigmentadas, que pueden persistir varios meses. Si el cuadro se desarrolla en el espesor de la dermis, aparecerán las cicatrices, deprimidas y de pequeño tamaño, o puede llegar configurar auténticos queloides, que pueden calcificarse (García et al., 2014).
Todas estas lesiones se pueden encontrar en el acné vulgar –también llamado polimorfo por este motivo– en diferente número, configurando variantes según el predominio de unas u otras. Así, en la forma clínica más abundante de la patología podemos diferenciar entre los siguientes tipos: i) acné vulgar comedoniano; ii) acné vulgar papulopustuloso (Figura 2); c) acné vulgar noduloquístico (Figura 3); y acné vulgar cicatricial. También se puede definir la forma clínica del acné teniendo en cuenta el mayor o el menor número de comedones frente a las lesiones inflamadas, llamándose en el primer caso acné no inflamatorio y, en el segundo, acné inflamatorio.
En cualquier caso, se distinguen grados progresivos, en función de la gravedad. Una de las escalas más empleadas para valorar clínicamentela gravedad del acné es la escala de Valoración Global del Investigador (IGA, por sus siglas en inglés) para la cara, y la Valoración Global por el Médico (PGA, por sus siglas en inglés) para el tronco; ambas determinan 5 grados (AEMPS, 2019):
Grado 0: pre-acné. Piel limpia sin lesiones inflamatorias o no inflamatorias.
Grado 1: leve. Unos cuantos comedones dispersos y unas cuantas pequeñas pápulas.
Grado 2: medio. Fácilmente reconocible, con menos de la mitad de la superficie involucrada; algunos comedones y algunas pápulas y pústulas.
Grado 3: moderado. Más de la mitad de la superficie afectada. Muchos comedones, pápulas y pústulas; puede haber algún nódulo presente.
Grado 4: grave. Toda la superficie involucrada. Cubierta con comedones, numerosas pápulas y pústulas; puede haber algunos nódulos presentes.
Normalmente, un mismo enfermo puede presentar más de un tipo de lesión, lo que hace que se establezca un cierto solapamiento entre los distintos grados.
Desde una perspectiva clínica, se han descrito otras formas de acné menos comunes:
Acné neonatal: se caracteriza por la presencia de comedones cerrados en las mejillas, siendo infrecuentes las pápulas y pústulas. Suele aparecer en los primeros meses de vida y parece ser debido al paso transplacentario de andrógenos desde el organismo materno. No requiere ningún tratamiento.
Acné cosmético: es una forma leve aunque persistente, que suele aparecer en mujeres de mediana edad. Es típica la presencia de comedones cerrados de escasa actividad inflamatoria. Suele ser debido a la aplicación de cremas con lanolina o manteca de cacao; con el ánimo de esconder el proceso, suelen usarse más cosméticos con estos ingredientes, lo que tiende a cronificar el cuadro.
Acné conglobata: es una forma severa que suele afectar a los varones, en la que son característicos los comedones dobles o triples, los nódulos, los quistes y, en ocasiones, grandes abscesos fistulizados y muy exudativos; puede dejar como secuela importantes cicatrices queloides. Tiene un curso crónico y recidivante, que llega a afectar al tórax, la espalda, los hombros y, en ocasiones, las nalgas. El pioderma facial es una variante que suele aparecer en mujeres, con nódulos eritematosos en la cara, de inicio súbito, sin comedones. Se asocia con frecuencia a episodios de tensión emocional, pudiendo permanecer en actividad hasta 2 años.
Acné queloidiana: se manifiesta en la nuca en varones y cursa con cicatrices queloideas en esta región, como resultado de la inflamación crónica y recidivante.
Acné fulminans: es la forma más infrecuente, quizá algo más común en varones. Constituye una forma grave que tiende a formar úlceras y cicatrices, e incluso puede cursar con afectación del estado general del organismo, con fiebre, artralgias, leucocitosis y aumento de la velocidad de sedimentación sanguínea.
Hay también reacciones acneiformes, que recuerdan al acné pero que no tienen el mismo curso ni origen, ya que frecuentemente son debidas a estímulos irritantes sobre la piel. Este es el caso típico asociado al empleo de ciertos productos en la piel, tales como jabones, detergentes, filtros solares o cosméticos; a veces, pueden aparecer como manifestaciones cutáneas de enfermedades sistémicas, como, por ejemplo, el síndrome de Cushing, alteraciones adrenogenitales, casos de virilización, etc. Por otra parte, el acné ocupacional aparece por la acción de múltiples productos industriales, como los alquitranes de hulla y los hidrocarburos, entre otros. Dentro de este grupo están también los casos de acné producidos por estímulos mecánicos o que aparecen en zonas de roce o de presión, como en la barbilla de los violinistas.
En general, el sol tiende a mejorar el acné, independientemente del tipo, aunque existen formas raras que aparecen, se desarrollan o empeoran en verano o en primavera, desapareciendo en otoño; esos pacientes suelen presentar lesiones localizadas en las zonas expuestas al sol, como rostro, torso o espalda. En otras ocasiones, son los trastornos psiquiátricos los que llevan al sujeto a una permanente manipulación de las lesiones que hace imposible la total curación del cuadro; e incluso permite el desarrollo de grandes cicatrices.
En cuanto a su epidemiología, se trata de la patología más frecuente de las que afectan a la piel, ya que entre el 80% y el 90% de las personas la padece en mayor o menor grado a lo largo de su vida, suponiendo hasta el 25% de las consultas al dermatólogo. En Europa, se estima que hay aproximadamente 70 millones de afectados y se cree que afecta al 9,4% de la población mundial. En España, la prevalencia del acné entre la población escolar de entre 12 y 18 años se estima en un 74% (esto es, casi 3 de cada 4 adolescentes), sin diferencias estadísticamente significativas en cuanto al sexo, encontrándose la máxima prevalencia entre los 14 y los 16 años de edad. Se cree que en torno a la mitad de los pacientes con acné facial padece también manifestaciones en el tronco superior. La evolución del acné es, en general, de varios años de duración y puede dar lugar a una remisión espontánea. Aunque muchos pacientes mejoran sus lesiones alrededor de los 20 años, algunos presentan acné hasta la tercera o cuarta década de sus vidas (algunos estudios apuntan que hasta el 22% de las mujeres adultas lo padecen). No obstante, el pronóstico del acné es generalmente favorable.
Tiene una etiopatogenia compleja, en la que influyen la genética, alteraciones en la glándula sebácea, colonización por bacterias y los andrógenos. La teoría patogénica más aceptada considera el acné como un fenómeno secuencial, en el que los andrógenos inducen una producción de sebo (secreción lipídica) excesiva, que, junto a una anormal descamación –hipercornificación– del epitelio del folículo pilosebáceo, provoca obstrucción, circunstancia en la que se favorece la multiplicación bacteriana, especialmente del bacilo gram positivo y anaerobio Propionibacterium acnés, causante principal de la inflamación (dermatosis).
Los andrógenos (testosterona, androstenediona, dihidroepiandrosterona, etc.) se transforman en la piel en testosterona, que en las glándulas sebáceas, mediante la 5-α-reductasa, se transforma en dihidrotestosterona (DHT), más potente, que va a estimular la actividad metabólica y proliferativa de la glándula sebácea. También hormonas hipofisarias, como la ACTH y la TSH, tienen una influencia indirecta, al inducir la 5-α-reductasa. Las lipasas bacterianas van a incrementar la presencia de ácidos grasos libres, irritantes para el folículo; y, junto con la actividad de proteasas y hialuronidasas, van a contribuir a modificar la queratinización del mismo, iniciando el desarrollo de la lesión característica del acné: el comedón. El Propionibacterium genera, además, sustancias quimiotácticas y análogos de mediadores inflamatorios que van a desencadenar el proceso. Se considera que el acné en cara y en trono tienen una fisiopatología similar.
Más allá de la predisposición del sujeto, existen distintas sustancias que pueden favorecer su aparición. Así, se ha se ha descrito la aparición de reacciones acneiformes tras el tratamiento con diversos fármacos, tanto de administración tópica como sistémica, tales como: tratamientos hormonales, antituberculosos, antiepilépticos, sales de litio, tratamientos inmunosupresores, corticoides3 , y hasta la propia terapéutica antiacné.
Tratamiento
Los regímenes de tratamiento farmacológico deben iniciarse de manera temprana, especialmente para impedir la formación de cicatrices (que, en caso de producirse, también deben ser consideradas en el tratamiento). Está fundamentalmente encaminado a corregir los factores etiológicos que contribuyen a su desarrollo, regulando la secreción sebácea, evitando la obstrucción del folículo y la formación del comedón y disminuyendo la población bacteriana. Para ello se utilizan fármacos con una o varias actividades farmacológicas: antimicrobiana, queratolítica, antiseborreica y antiinflamatoria. Se suele realizar un tratamiento continuado y escalonado, comenzando habitualmente con tratamientos tópicos. Debe evitarse la manipulación de las lesiones, no es necesario modificar los hábitos alimentarios (no hay evidencias de que varíen la gravedad del proceso) y se debe aconsejar una higiene específica adecuada.
El ácido azelaico, disponible en crema al 20% o en gel al 15%, tiene propiedades antimicrobianas y comedolíticas. Además, es un inhibidor competitivo de la tirosinasa y disminuye de así la pigmentación; este efecto puede utilizarse en los pacientes con hiperpigmentación posinflamatoria. Por lo general, es bien tolerado, aunque puede producir una sensación de ardor transitoria, y es seguro durante el embarazo.
El peróxido de benzoílo es un fármaco habitualmente prescrito por los dermatólogos para el tratamiento del acné. Se trata de un poderoso agente antimicrobiano que también es queratolítico. Está disponible en forma de geles a una concentración de entre el 2,5 y el 10%, pudiendo producir una mayor sequedad e irritación a mayor concentración (la tolerancia aumenta si se emplea micronizado). Su actividad antibacteriana se atribuye a la liberación de radicales libres de oxígeno, que oxidan las proteínas bacterianas, lo que hace que carezca de riesgo de resistencias bacterianas y sea un agente sea ideal para la terapia de combinación con otros fármacos antimicrobianos.
Por otra parte, los antibióticos tópicos actúan únicamente sobre las lesiones inflamatorias superficiales. Al reducir la carga bacteriana (y, por tanto, la liberación de ácidos grasos libres por acción de sus lipasas), ejercen también una acción antiinflamatoria. Los principales antibióticos tópicos empleados en el tratamiento del acné son la clindamicina al 1% y la eritromicina al 2%, los cuales también se han usado en presentaciones combinadas con peróxido de benzoílo en una combinación que mejora el perfil de resistencias bacterianas (sobre todo, de P. acnes). La mejoría clínica con estos antibióticos suele darse en torno a las 6 semanas de tratamiento. El nadifloxacino es una fluoroquinolona que surge como alternativa a la eritromicina y la clindamicina en caso de resistencias.
Los retinoides tópicos son otro grupo de fármacos (derivados naturales o sintéticos de la vitamina A) ampliamente usados frente al acné vulgar. Actúan modificando la pared del conducto pilosebáceo y determinando una mayor dehiscencia de la queratina, responsable de la obstrucción del conducto. Básicamente, se utilizan la tretinoína al 0,025-0,1% y el adapaleno al 0,1%; la isotretinoína al 0,05% ha caído en desuso por vía tópica, y el tazaroteno, disponible en otros países, no está autorizado en España para el tratamiento del acné. Las propiedades comedolíticas y antiinflamatorias de los retinoides tópicos los hacen ideales para la terapia de mantenimiento del acné. Pueden causar irritación como efecto secundario (el que menor riesgo comporta es el adapaleno), por lo que los pacientes deben cuidarse de la exposición al sol y usar una pantalla solar; además, por su carácter teratógeno, debe aconsejarse adecuadamente a las pacientes en edad fértil sobre la adopción de medidas anticonceptivas.
En casos de acné moderado a grave estaría indicada la terapia sistémica. En este caso pueden emplearse antibióticos, hormonas y retinoides. Los antibióticos más utilizados son las tetraciclinas, aunque con frecuencia se necesitan varias semanas de tratamiento. Entre los macrólidos, se utiliza la eritromicina, especialmente en mujeres embarazadas y niños, pero también se emplea la azitromicina.
El tratamiento hormonal del acné tiene como objetivo contrarrestar los efectos de los andrógenos sobre las glándulas sebáceas. Los antiandrógenos, como el acetato de ciproterona, bloquean los receptores androgénicos de la glándula sebácea, disminuyendo la producción de sebo. Por su acción feminizante, el acetato de ciproterona solo tiene indicación en la mujer a dosis de 50-100 mg/día; debe emplearse en asociación con un estrógeno, como el etinilestradiol a dosis de 0,03-0,05 mg/día, para conseguir una acción anticonceptiva (pues el acetato de ciproterona es teratógeno) y para minimizar los efectos secundarios sobre el ciclo menstrual.
Los corticoides son también beneficiosos en las formas intensas y graves. Las dosis deben ser bajas, pues en dosis altas son comedogénicos y dan lugar a erupciones acneiformes. La corticoterapia suele utilizarse por periodos de tiempo limitado y son frecuentes las recidivas postratamiento.
Por último, en cuanto al tratamiento con retinoides sistémicos, el efecto fundamental de la isotretinoína en el tratamiento del acné intenso consiste en una reducción de la secreción sebácea, que se relaciona no con el bloqueo androgénico sino con una atrofia casi completa de las glándulas sebáceas. Además, disminuye la cohesión de los queratinocitos del orificio folicular, favoreciendo la eliminación de los comedones, y disminuye la población de P. acnes, ejerciendo también un efecto antiinflamatorio. Está aprobado, y es quizá el fármaco sistémico más usado, para el tratamiento de pacientes con acné nodular grave y refractario, aunque con frecuencia se utiliza en otros tipos de acné moderado-grave que no responden a otros tratamientos.
Con isotretinoína por vía oral (a dosis oscilante entre 1 y 0,05 mg/kg/día, siendo esta última mucho mejor tolerada) suele alcanzarse una remisión completa en casi todos los casos y la duración de esta remisión se prolonga durante meses o varios años en la gran mayoría de pacientes (70-90%), por lo que su introducción supuso un importante avance. Como efectos secundarios, que son dosis-dependientes, se incluyen, entre otros, sequedad de piel y mucosas, con queilitis en el 100% de los casos, elevación de los lípidos sanguíneos, alteraciones oculares, cefaleas, riesgo de depresión, náuseas y mialgias. Pero el aspecto de seguridad más importante es la teratogenicidad, influyendo sobre la organogénesis, lo cual limita su uso en la mujer fértil, debiendo, si llega a administrársele, tener un estricto control anticonceptivo (y también del hemograma y funcionalidad hepática) durante el tratamiento y hasta un mes después de suspenderlo.
Acción y mecanismo
El trifaroteno es un derivado de retinoide que actúa como un potente agonista del receptor γ del ácido retinoico (RARγ); se caracteriza por una especificidad muy elevada por este receptor, uniéndose con una afinidad 50 veces mayor que sobre RARα y 8 veces mayor que sobre RARβ4 ; no presenta, además, actividad sobre receptores X de retinoides (RXR). Conviene recordar que todos los subtipos de receptores de ácido retinoico (RAR) son receptores nucleares hormonales cuya activación provoca su heterodimerización con los RXR formando complejos que reconocen regiones específicas del ADN (elementos de respuesta a retinoides) en las regiones promotoras de genes diana, implicados en el crecimiento celular, la diferenciación y la apoptosis, cuya expresión modulan. Aunque el proceso exacto por el cual mejora el acné no se conoce completamente, el medicamento ha sido autorizado para el tratamiento cutáneo de acné vulgar en la cara, en el pecho y/o en la espalda en pacientes mayores de 12 años, en presencia de numerosos comedones, pápulas y pústulas.
Al ser el RARγ el receptor de retinoides de largo más abundante en la piel, su estimulación resulta en una modulación de la expresión de los genes diana de los retinoides –relacionados con diversos procesos que incluyen la diferenciación y proliferación celular, respuesta al estrés, apoptosis y cascadas antiinflamatorias– en los queratinocitos inmortalizados (en los que es estable más de 24 h) y en la epidermis reconstruida. Los estudios con cultivos de piel humana ex vivo, los modelos in vivo y los ensayos clínicos (administración tópica durante 4 semanas) también demostraron que modula la expresión de genes relacionados con nuevas rutas bioquímicas involucradas en el acné, como la proteólisis, la adhesión celular o la hidratación de la piel.
En modelos murinos (rino-ratón), el fármaco ha demostrado una marcada actividad comedolítica (reducción del 98% de los comedones), reduciendo numerosos comedones y aumentando significativamente el grosor de la dermis. En dicho modelo, el trifaroteno produjo el mismo efecto comedolítico que otros retinoides conocidos (como tazaroteno o la tretinoína), con una concentración 10 veces menor. Además, ha demostrado tener una prometedora actividad antiinflamatoria y despigmentante (efecto visible tras 6 semanas de administración), y ser rápidamente metabolizado en microsomas hepáticos humanos, lo que permite predecir un perfil toxicológico favorable debido a bajos niveles sistémicos. Esto, unido a que la ausencia de selectividad sobre los receptores RARβ dérmicos se ha relacionado con una posible reducción de la irritación cutánea, sugiere que el balance eficacia/seguridad de trifaroteno puede ser mejor que el de otros retinoides tópicos no selectivos (Aubert et al., 2018).
Aspectos moleculares
En términos estructurales, los retinoides se clasificaban, hasta ahora, en 3 generaciones: el retinol, la tretinoína, la alitretinoína y la isotretinoína pertenecen a la primera generación, etretinato y acitretina a la segunda (retinoides aromáticos, generados por modificaciones en el grupo cíclico terminal), y tazaroteno y adapaleno a la tercera (estos dos, llamados arotinoides, incluyen más modificaciones moleculares que añaden a la actividad comedolítica las propiedades antiinflamatorias); algunos autores indican que adapaleno –derivado del ácido naftoico– no encaja en esa tercera generación. Para el tratamiento del acné se han autorizado los de primera y tercera generación, todos los cuales son agonistas duales de RARβ y RARγ (los de primera generación también de RARα); sus propiedades antiacneicas se han atribuido a efectos antiproliferativos y antiinflamatorios y a una normalización de la diferenciación de keratinocitos.
Diversos estudios de cristalografía de rayos X de los receptores RAR permitieron describir las partes de las moléculas que conferirían agonismo selectivo hacia un subtipo concreto de receptor. Así, se postuló que los agonistas selectivos para RARγ establecerían una interacción mediante puente de hidrógeno con el grupo sulfidrilo de la metionina en posición 272 del receptor (en esa posición los receptores RARα y β tienen un residuo de isoleucina) o bien gracias a la flexibilidad intrínseca de dicho residuo de metionina, que permite interacciones en el “bolsillo” generado con su movimiento. En base a estas y otras observaciones, Thoreau y colaboradores (Thoreau et al., 2018) desarrollaron un diseño racional de síntesis de una serie de moléculas triarílicas con modificaciones estructurales diversas que fueran potencialmente selectivas para RARγ, desembocando en la selección de trifaroteno –entre 30 candidatos– para su estudio clínico.
Trifaroteno es un derivado del ácido terfenílico químicamente estable. Se trata de un retinoide de cuarta generación que conserva cierta similitud estructural con los retinoides de tercera generación usados frente al acné (en mayor medida con adapaleno), más distante de los de primera generación (Figura 4). El nombre químico de trifaroteno es el ácido 3”-tertbutil-4’-(2-hidroxi-etoxi)-4”-pirrolidin-1-il-[1,1’,3’,1”]terpenil-4-carboxílico, que se corresponde con la fórmula C29H33 NO4 y el peso molecular de 459,58 g/mol. Se presenta como un polvo blanco o blanquecino (o ligeramente amarillento) con una temperatura de fusión de 245ºC y prácticamente insoluble en agua.
Eficacia y seguridad clínicas
La eficacia y seguridad clínicas de trifaroteno por vía tópica han sido adecuadamente contrastadas en la indicación y dosis autorizadas mediante dos ensayos pivotales de fase 3 de idéntico diseño (PERFECT1 y PERFECT2): aleatorizados (1:1), doble ciego, multicéntricos y multinacionales, de grupos paralelos y controlados con crema vehículo. En conjunto, evaluaron la farmacodinamia clínica del fármaco en un total de 2.420 pacientes de más de 9 años de edad (media de 19,6 años) con acné vulgar facial y troncal moderado, definido como grado 3 según las escalas IGA para el acné facial y PGA para el acné en tronco. La mitad de ellos fueron tratados con una aplicación diaria de trifaroteno durante un máximo de 12 semanas, periodo durante el cual podían utilizar un agente hidratante de su elección, pero siempre dejando un intervalo de 1 hora antes y después de la aplicación del fármaco experimental.
Las características demográficas y de la patología de los pacientes estaban bien balanceadas entre los distintos brazos en los dos estudios. A grandes rasgos, un 87% del total de los pacientes eran de raza blanca (caucásicos), el 55% eran mujeres, y los grupos etarios principales eran los adolescentes de entre 12 y 17 años (47%) y los adultos de ≥ 18 años (52%); solo un 1,4% de los pacientes incluidos correspondían a la población pediátrica de entre 9 y 11 años. Al inicio del estudio, los pacientes presentaban un promedio de 36 lesiones inflamatorias (rango 7-200) y 52 lesiones no inflamatorias (rango 21-305) en la cara, y una media de 38 lesiones inflamatorias (rango 0-220) y 46 lesiones no inflamatorias (rango 0-260) en el tronco. Se excluyeron pacientes con formas severas o secundarias de acné.
La eficacia del tratamiento se determinó, a la semana 12, mediante tres variables co-primarias en ambos estudios: la tasa de éxito en base a una mejora de 2 grados en la escala IGA (proporción de pacientes con piel facial aclarada o casi aclarada, es decir, grados 0 o 1), el cambio absoluto y porcentual respecto al estado basal en el recuento de lesiones inflamatorias y en el recuento de lesiones no inflamatorias de acné facial. Como variables secundarias de eficacia se consideraron la tasa de éxito determinada por una mejora de al menos 2 grados respecto al estado basal en el acné en tronco según la escala PGA (grados 0 o 1), así como el cambio absoluto y porcentual respecto al estado basal en el recuento de lesiones inflamatorias y no inflamatorias en esa localización.
Los principales resultados divulgados, derivados del análisis por intención de tratar e imputación múltiple, se recogen en la siguiente Tabla 1 (Tan et al., 2020). Los valores de todas las variables alcanzaron significación estadística a la semana 12 a favor de la crema de trifaroteno frente a la de vehículo; no obstante, esa superioridad significativa era ya evidente desde las semanas 4 a 8 para las tasas de éxito, e incluso antes (semanas 1 a 4) para la reducción de lesiones acneicas en cara y tronco.
Conviene destacar que, debido al bajo número de pacientes pediátricos entre 9 y 11 años que fueron evaluados en los ensayos de fase 3 (19 en PERFECT1 y 15 en PERFECT2), no pudo demostrarse de forma sólida la eficacia de trifaroteno en esa subpoblación. Sin embargo, los análisis por subgrupos de los datos obtenidos confirman que la eficacia del fármaco a las 12 semanas es consistente en pacientes adolescentes (12-17 años), cuyo número fue bastante mayor (573 en PERFECT1y 555 en PERFECT2). En esos pacientes, las tasas de éxito del tratamiento con trifaroteno para el acné facial –escala IGA– se situaron entre el 25,6-35,8% (vs. 14,7-20,4% con crema vehículo), y para el acné en tronco –escala PGA– entre el 31,8-38,7% (vs. 21,0-25,8%). De igual modo, en la comparación con la eficacia de la crema vehículo, la reducción media absoluta tanto de lesiones inflamatorias como de lesiones no inflamatorias en acné facial (diferencia media de -3,8 a -5,3 lesiones inflamatorias y de -9,6 a -11,0 lesiones no inflamatorias) y en acné de tronco (diferencia media de -3,4 a -6,2 lesiones inflamatorias y de -5,0 a -5,7 lesiones no inflamatorias) alcanzó significación estadística (p < 0,001).
Por otro lado, la seguridad clínica de trifaroteno por vía tópica, definida fundamentalmente a partir de los datos de los ensayos pivotales (en que recibieron el fármaco 1.220 pacientes durante 12 semanas), se caracteriza por frecuentes eventos adversos cutáneos que aparecen localmente en la zona de aplicación, la mayoría pasajeros y gravedad leve-moderada5. En la cara, se notificaron reacciones cutáneas leves, moderadas y graves en hasta un 39%, 29,7% y 6,2% de los pacientes, respectivamente; y en el tronco, en un 32,9%, 18,9% y 5,2% de los pacientes, respectivamente. La gravedad máxima se alcanzó habitualmente en la semana 1 en la cara y de la semana 2 a 4 en el tronco, disminuyendo con el uso continuado del fármaco.
La incidencia de signos y síntomas de tolerabilidad local durante el tratamiento difirió ligeramente según la zona de aplicación del fármaco. En el caso del acné facial, destaca la frecuencia de eritema (24-33%, grave en el 3-10% de pacientes), descamación (21-33%, grave en 3-7%), sequedad (23-36%, grave en 3-7%) y ardor/quemazón (16-25%, grave en 4-7%); en el tronco, se observó una menor frecuencia de eritema (15-23%, grave en el 3-7% de pacientes), descamación (11-17%, grave en 0-3%), sequedad (11-21%, grave en 1-3%) y ardor/quemazón (9-13%, grave en 3-6%). Las reacciones adversas más frecuentes relacionadas con trifaroteno fueron irritación en el lugar de aplicación (7,5% vs. 0,3% con crema vehículo), prurito en el lugar de aplicación (2,4% vs. 0,8%) y quemaduras solares (2,6% vs. 0,5%). En cualquier caso, parece un fármaco bien tolerado, habida cuenta de que las tasas de discontinuación por eventos adversos (sobre todo, irritación cutánea y dermatitis alérgica) fueron muy bajas (< 2%) entre los pacientes de los brazos de tratamiento.
Estudios previos en el desarrollo clínico del fármaco demostraron que la absorción sistémica del fármaco es muy baja, incluso cuando se aplica diariamente a concentraciones más elevadas de las autorizadas (Wagner et al., 2020). Esto explica que en los ensayos pivotales no se observaran cambios relevantes en los signos vitales (incluyendo función cardiovascular), en parámetros de laboratorio ni en el examen físico de los pacientes. También se descartó una posible interacción farmacológica con el uso de anticonceptivos orales en mujeres susceptibles de embarazo. No obstante, aunque la EMA considera que la absorción sistémica de retinoides por vía tópica es a priori residual, mantiene la contraindicación de su uso, por el riesgo de teratogenicidad, en mujeres embarazadas o que estén planificando un embarazo, al no haberse estudiado rigurosamente los efectos de estos fármacos por vía tópica sobre el embarazo (EMA, 2018).
Finalmente, hay que destacar los resultados de un reciente ensayo clínico que ha evaluado la eficacia y seguridad de trifaroteno a más largo plazo, en tratamiento de 1 año de duración (Blume-Peytavi et al., 2020). Este estudio de fase 3, multicéntrico y multinacional, abierto y no controlado (no comparativo), incluyó también pacientes (N= 453) de ≥ 9 años de edad (media de 18,3 años) con acné vulgar facial o troncal moderado (grado 3). Todos ellos fueron tratados con una aplicación diaria de trifaroteno a fin de evaluar la tolerabilidad local, la seguridad y la eficacia del tratamiento, así como la influencia sobre la calidad de vida. Un total de 376 pacientes (83%) completaron al menos 6 meses de tratamiento y 342 (75,5%) finalizaron el año de tratamiento.
Los resultados de seguridad y eficacia a 1 año confirman los hallazgos de los estudios pivotales. Se reportaron eventos adversos relacionados con el fármaco en el 12,6% de pacientes, ninguno de ellos severo, asociados a una tasa de discontinuación de < 3% de pacientes. La práctica totalidad eran reacciones cutáneas (prurito – 4,6%, irritación – 4,2%, y quemaduras solares – 1,8%) que ocurrieron durante los 3 primeros meses; la incidencia de irritación local aumentó durante la primera semana en la cara y hasta las semanas 2 a 4 en el tronco, decreciendo posteriormente. En términos de eficacia, el tratamiento con trifaroteno confirmó la mejoría clínica del acné, que fue más relevante al año de tratamiento en comparación con periodos más cortos: la tasa de éxito en la escala IGA (acné facial) aumentó desde el 26,6% en la visita de la semana 12 hasta el 65,1% en la visita de la semana 52, y la tasa de éxito en la escala PGA (acné troncal), desde el 38,6% en la visita de la semana 12 hasta el 66,9% en la visita de la semana 52; además, el éxito global (en IGA y PGA) experimentado por el mismo sujeto se incrementó desde un 22,0% en la semana 12 hasta un 57,9% en la semana 52. Además, el 54% de los pacientes que completaron el tratamiento de 1 año reportaron valores de 0 a 1 en un cuestionario validado de calidad de vida dermatológica (DLQI) –indicativo de que el acné no afectaba a su calidad de vida–, en comparación con el 22,6% de los pacientes en la visita inicial.
Aspectos innovadores
El trifaroteno es un nuevo derivado de retinoide que actúa como un potente agonista del receptor γ del ácido retinoico (RARγ), con una eficacia de activación y una especificidad muy elevadas por este receptor, uniéndose con una afinidad 50 veces mayor que sobre RARα y 8 veces mayor que sobre RARβ; no presenta, en cambio, actividad sobre receptores X de retinoides (RXR). Así, habida cuenta que los RARγ son los receptores de retinoides más abundantes en la piel, trifaroteno será capaz de modular la expresión de genes diana implicados en el crecimiento y la diferenciación celular, la respuesta al estrés, cascadas antiinflamatorias y la apoptosis, entre otros procesos. Si bien el proceso exacto por el cual mejora el acné no se conoce completamente, el medicamento ha sido autorizado para el tratamiento cutáneo (en forma de crema) de acné vulgar en la cara, en el pecho y/o en la espalda en pacientes mayores de 12 años, en presencia de numerosos comedones, pápulas y pústulas.
La evidencia de la eficacia y seguridad de trifaroteno conducente a su aprobación deriva de dos amplios ensayos clínicos de fase 3 de idéntico diseño: doble ciego, de grupos paralelos y controlado por crema vehículo. Estos aleatorizaron un total de 2.420 pacientes de ≥ 9 años de edad con acné vulgar facial y troncal moderado (grado 3 según las escalas IGA para el acné facial y PGA para el acné en tronco). A las 12 semanas de tratamiento con una aplicación diaria, la crema de trifaroteno fue significativamente superior al vehículo en los dos estudios, tanto respecto a las tasas de éxito terapéutico como a la reducción de las lesiones acneicas en cara (variables co-primarias) y en tronco (variables secundarias).
Así, la tasa de éxito en acné facial según la escala IGA fue de 29,4% y 42,3% en los dos ensayos (vs. 19,5% y 25,7% para el vehículo; p< 0,001), mientras que la reducción del recuento de lesiones inflamatorias fue de 19,0 y 24,2 (vs. 15,4 y 18,7; p< 0,001) y la de lesiones no inflamatorias de 25,0 y 30,1 (vs. 17,9 y 21,6; p< 0,001). Adicionalmente, las variables secundarias confirman la eficacia de trifaroteno en acné en tronco, con una tasa de éxito según la escala PGA de 35,7% y 42,6% en los dos ensayos (vs. 25,0% y 29,9% para el vehículo; p< 0,001), una reducción del recuento de lesiones inflamatorias de 21,4 y 25,5 (vs. 18,8 y 19,8; p< 0,001) y de lesiones no inflamatorias de 21,9 y 25,9 (vs. 17,8 y 20,8; p< 0,001). De manera interesante, el beneficio clínico con trifaroteno presentó un inicio rápido, evidenciado por la reducción de las lesiones acneicas desde la primera y la segunda semana en la cara y en el tronco, respectivamente; las tasas de éxito superaban a las del vehículo entre las semanas cuarta (acné facial) y octava (acné troncal). A pesar de que el análisis por subgrupos de edad no pudo confirmar la eficacia de trifaroteno en pacientes pediátricos de entre 9 y 11 años (que se excluyeron de la indicación autorizada) por el bajo número de participantes, la magnitud del efecto fue similar en adultos y en población adolescente (entre 12 y 17 años).
Cabe destacar, además, que un ensayo clínico abierto de un solo brazo a largo plazo confirmó la consistencia del continuo beneficio clínico con una administración diaria de trifaroteno: 342 pacientes completaron 1 año de tratamiento, en quienes se confirmaron tasas de éxito crecientes con el tiempo, que alcanzan valores del 65,1% para el acné facial (vs. 26,6% en la semana 12) y del 66,9% para el acné troncal (vs. 38,6% en la semana 12). Se verificó también una marcada mejoría temporal en el mismo paciente, de manera que se reportaban altas tasas de satisfacción con el tratamiento prolongado; más de la mitad de los pacientes (54%) confirmaba al año de tratamiento que el acné no afectaba a su calidad de vida.
En términos de seguridad, trifaroteno por vía tópica ha sido adecuadamente caracterizado con datos de más de 1.500 pacientes que han recibido el fármaco durante al menos 3 meses. Presenta un perfil toxicológico aceptable (en línea con el conocido para otros retinoides clásicos), con tasas de discontinuación bajas (< 2%), y clínicamente manejable con ajustes posológicos o el uso de hidratantes no comedogénicos o limpiadores suaves. Se caracteriza por la aparición de signos y síntomas locales leves-moderados (eritema, descamación, sequedad y ardor/quemazón) de incidencia variable entre el 10 y el 30%, que aparecen con mayor gravedad en las primeras semanas de tratamiento para luego disminuir; la tolerabilidad fue mejor en tronco que en cara. Las reacciones adversas más frecuentemente relacionadas con trifaroteno fueron irritación (7,5% vs. 0,3% con vehículo) y prurito en el lugar de aplicación (2,4% vs. 0,8%) y quemaduras solares (2,6% vs. 0,5%). Los resultados del estudio de seguridad a largo plazo concuerdan y confirman los hallazgos de los ensayos pivotales. Además, su escasa absorción sistémica y su rápido metabolismo hepático hacen que carezca de eventos adversos sistémicos, lo que permite su uso en amplias áreas cutáneas del tronco; sin embargo, se mantiene la contraindicación de su uso en embarazo (o planificación del mismo) por el riesgo de teratogenicidad propia de los retinoides.
Aunque los estudios pivotales se ven limitados por la ausencia de regímenes posológicos adecuadamente establecidos y por la no evaluación de tratamientos tópicos o sistémicos adyuvantes (por ejemplo, la combinación de trifaroteno con peróxido de benzoilo o antibióticos tópicos, que en principio podría ser más eficaz), el principal hándicap es la ausencia de comparador activo. Ello impide posicionar trifaroteno frente a otras alternativas de tratamiento tópico del acné en adolescentes y adultos y, en particular, frente a los otros retinoides como tretinoína y adapaleno, disponibles desde hace décadas.
Así pues, parece evidente que en un futuro se requieren ensayos clínicos que comparen directamente la eficacia y tolerabilidad de trifaroteno con los retinoides clínicamente establecidos u otras opciones como peróxido de benzoilo o antibióticos. Hasta entonces, el nuevo fármaco podría situarse como una alternativa más en primera línea del tratamiento tópico del acné moderado. Algunos autores apuntan a que no está muy claro cuál de los retinoides es más eficaz. Una comparación indirecta no ajustada (que carece de robustez sacar conclusiones) a la vista de los resultados de eficacia de tretinoína y adapaleno sugiere que trifaroteno aporta un beneficio clínico similar o ligeramente superior. Por ejemplo, un meta-análisis de los datos de 900 pacientes demostró una reducción media de las lesiones acneicas en un 57% con adapaleno y en un 53% con tretinoína, ambos en monoterapia; adapaleno demostró un inicio del efecto más rápido y mejor tolerabilidad cutánea (Kassir et al., 2020). Con trifaroteno, la reducción de lesiones acneicas en cara y tronco se sitúa en el rango de 49-66%.
Aunque no representa una innovación importante en términos mecanísticos, la principal novedad que aporta trifaroteno, y que lo diferencia del resto de retinoides tópicos (agonistas duales de RARβ y RARγ), en su selectividad por el receptor mayoritario en la piel, RARγ. Si bien no está del todo clara la significación clínica de dicha selectividad, el reducido agonismo sobre los receptores RARβ dérmicos podría relacionarse con una menor incidencia de la irritación cutánea (Bell et al., 2020); en todo caso, esa mejor tolerabilidad teórica debe ser comprobada en estudios comparativos.
Conviene destacar que los aquí comentados son los primeros estudios comparativos y bien diseñados que, hasta la fecha, han evaluado rigurosamente un tratamiento del acné del tronco (hombros, espalda y pecho), que afecta a una amplia proporción de pacientes con acné facial (> 50%) y se considera una necesidad médica no cubierta, habiendo sido tratado hasta la fecha con las mismas alternativas usadas en acné facial, a pesar de la falta de evidencia. En definitiva, estamos ante el primer retinoide (de cuarta generación) desarrollado en las últimas dos décadas, siendo el tercero aprobado en España para el tratamiento del acné por vía tópica (tras tretinoína y adapaleno) y el primero que ha demostrado eficacia notable en ensayos clínicos frente al acné en tronco. No representa una cura de la patología pero permite un buen control de los síntomas con una tolerabilidad aceptable.
Múltiples virus pueden infectar la piel de los seres humanos, manifestándose como diversas patologías según edad, género y estado inmunológico del huésped. Las lesiones cutáneas pueden ser el resultado de una reacción inmunitaria a distancia, sin que se llegue a demostrar directamente la presencia del virus en las lesiones (como sería el caso de enfermedades virales exantemáticas y las manifestaciones de la COVID-19), o bien estar producidas por el propio virus, siendo posible aislarlo en la piel, en cuyo caso produce lesiones potencialmente contagiosas (como el molusco contagioso o las verrugas vulgares).
El presente artículo de revisión se centra en las principales características clínicas –y terapéuticas– de las manifestaciones cutáneas por infecciones víricas más frecuentemente vistas en consulta de Atención Primaria, considerando su etiología y aportando una visión global para el diagnóstico clínico diferencial.
Introducción
Las manifestaciones cutáneas de las infecciones víricas representan un motivo frecuente de consulta en Atención Primaria.
Los virus son agentes infecciosos de estructura genómica sencilla, capaces de adueñarse del aparato sintético nuclear de la célula infectada, una vez que han conseguido penetrar en ella. Un grupo importante de virus tiene capacidad de replicar en células epiteliales, originando cuadros clínicos en los que se evidencia una destrucción celular o bien una hiperplasia del tejido infectado. La infección viral puede originar cambios estructurales en las células afectadas (por ejemplo, citolisis en herpes simple y zóster), infecciones crónicas activas (virus del papiloma humano y poxvirus), transformación neoplásica (virus del papiloma humano) o bien infección latente y recidivante (herpes simple y herpes zóster). Las lesiones cutáneas pueden producirse por inoculación directa (verrugas, moluscos), por invasión de la piel a partir de una infección sistémica (varicela) o por reactivación de virus latentes. En la mayoría de los casos las lesiones son autolimitadas.
Aunque existe una amplia variedad de virus que pueden provocar lesiones cutáneas, nos centraremos por su interés y frecuencia en las provocadas por herpesvirus, virus del papiloma humano, poxvirus y retrovirus.
Familia herpesviridae
Los virus herpes humanos (VHH) son virus ADN relativamente grandes, con replicación intranuclear, que produce típicas inclusiones intranucleares detectables en preparaciones histológicas tisulares. Siendo estructuralmente muy similares, se detecta una amplia variabilidad en cuanto a sus características bioquímicas y respuesta a fármacos antivíricos. Se hará hincapié a continuación en los aspectos clínicos del herpes simple y varicela zóster.
1) Herpes simple (VHS)
Es una de las infecciones más frecuentes que afectan al ser humano: hasta un 80% de la población presenta anticuerpos. Se produce por un virus perteneciente a la familia Herpesviridae, que presenta un núcleo de ADN bicatenario y proteínas, una cápside y una envoltura periférica derivada de la membrana nuclear de la célula infectada. El ser humano es el único reservorio y huésped natural.
Clásicamente, se distinguen dos tipos:
VHS tipo 1, responsable de la inmensa mayoría de infecciones orales, periorales, oculares o por encima de la cintura pelviana.
VHS tipo 2 o genital, que afecta a la zona anogenital y palmoplantar.
No obstante, según las prácticas sexuales, cualquiera de los dos tipos puede originar infección en cualquier territorio cutáneo-mucoso donde
se inocule.
Patogenia
En casi la totalidad de los casos, la primoinfección es asintomática y no suelen aparecer lesiones cutáneas. En esa situación, solo es demostrable mediante la realización de pruebas serológicas que demuestran la presencia de anticuerpos. Éstos no protegen frente a la reinfección o recurrencia ni siquiera por el mismo serotipo viral.
El VHS, tras su adsorción y penetración, pierde su envoltura y se incorpora a las proteínas del núcleo celular, comenzando a codificar la síntesis de enzimas replicadoras y el ARN mensajero. Tras la infección primaria (sintomática o asintomática) el virus migra través de los nervios sensitivos periféricos y se introduce en los ganglios de las raíces posteriores sensitivas donde entra en fase de latencia sin provocar muerte celular. Posteriormente, por motivos desconocidos, el genoma vírico se reactiva e inicia una nueva replicación. Los nuevos viriones se desplazan por los nervios periféricos llegando a la piel generando nuevas lesiones cutáneas.
Mecanismos de transmisión
Generalmente, se transmiten por contacto piel-piel, como es el caso del herpes gladiatorum en luchadores de lucha libre, o por contacto piel-mucosa, por el que el recién nacido puede infectarse en el canal del parto si la madre padece herpes genital. Factores como el déficit de higiene, hacinamiento, nivel social bajo, alteraciones inmunológicas, promiscuidad sexual y no guardar las precauciones suficientes para evitar el contacto con personas con brote herpético facilitan el contagio.
Manifestaciones clínicas del VHS
Primoinfeccion herpética. Deriva del primer contacto con el VHS. Aunque puede ser asintomática, suele afectar a niños, produciendo fiebre elevada, malestar general, vesículas en mucosa oral y adenopatías regionales: un cuadro clínico conocido como gingivoestomatitis herpética.
Herpes simple labial. Es la forma más frecuente de recurrencia del VHS tipo I: se presenta en el 38-45% de los individuos seropositivos para VHS. Se caracteriza por pródromos de dolor, ardor u hormigueo y presencia de vesículas agrupadas sobre una base eritematosa en el bermellón labial. Suele resolverse espontáneamente en 5-7 días, aunque el virus solo se excreta los 3 primeros días; en inmunodeprimidos, puede afectar a la lengua o el esófago, provocando un cuadro más severo.
Herpes simple genital. Aparecen en la piel vesículas arracimadas sobre una base eritematosa afectando a la región genital. Doloroso y con gran tendencia a las recidivas, puede tener importantes repercusiones psicológicas. En el sexo femenino no siempre es fácil distinguir las vesículas, por lo que hay que sospecharlo si existe dolor, prurito, disuria o leucorrea.
Panadizo herpético. Se produce por autoinoculación del virus al manipular con los dedos lesiones labiales, frecuente en niños, pediatras y odontólogos.
Eccema herpético o erupción variceliforme de Kaposi. Afectación herpética extensa en piel previamente afectada por otra patología cutánea de base. Se caracteriza por erosiones monomorfas y múltiples que se diseminan y vuelven hemorrágicas, costrosas, ulceradas y necróticas, con alto riesgo de sobreinfección; se trata de un proceso grave.
Herpes gladiatorum. Se debe a una contaminación piel-piel, que era frecuente entre gladiadores en la Grecia Clásica.
Queratoconjuntivitis herpética producida por VHS.
Herpes simple recidivante favorecido por una serie de factores responsables de la recurrencia, como son: fiebre, exposición solar, menstruación, ovulación, traumatismos, estrés, etc.
Herpes neonatal. Generalmente producido por el VHS tipo 2, afecta al neonato en ojos, boca y piel, comprometiendo al SNC hasta un 35% de los afectados y llegando a provocar –en un 20% de los casos– un herpes diseminado. Inicialmente, pueden presentar solo úlceras corneales o lesiones orales.
Diagnóstico
El diagnóstico de una infección por VHS es esencialmente clínico, ya que el paciente suele referir síntomas generales como astenia, dolor y malestar general. En la exploración física es característica la aparición de vesículas arracimadas sobre base eritematosa. Para confirmar la infección se puede realizar el estudio citológico del contenido de la vesícula (test de Tzanck). Otras técnicas útiles son la realización de una PCR específica para VHS o la detección de anticuerpos circulantes, si bien la serología solo se emplea para estudios de seroprevalencia y para identificar a individuos susceptibles. La mayoría de los estudios presentan una alta reactividad cruzada entre VHS-1 y VHS-2, por lo que se recomiendan técnicas como ELISA que identifiquen la IgG anti-glicoproteína G.
Tratamiento
En principio, se recomienda tratamiento tópico con soluciones astringentes, como sulfato de zinc o cobre al 1%, que pueden favorecer la desecación de las vesículas, evitando la sobreinfección.
Los antivirales de que disponemos hoy frente al VHS no son virucidas, no afectan a virus latentes, solo a los que están en fase de replicación, y producen resistencias en algunos pacientes como consecuencia de mutaciones en zimáticas (timidinkinasa). Se usan: aciclovir tópico al 5%, que solo es eficaz las 12 primeras horas y debe aplicarse cada 2-3 horas, y penciclovir tópico cada 4-6 horas, que también puede ser una alternativa si se usa desde etapas muy precoces.
Pero, por lo general, el herpes simple cura espontáneamente en un periodo de entre 5 y 7 días. El problema suelen ser las recidivas. En inmunocomprometidos y casos severos, se debe utilizar aciclovir intravenoso; en casos resistentes, se puede utilizar foscarnet o ciclofovir.
Por otra parte, en casos de herpes genital, se puede recurrir al tratamiento oral:
Primoinfección herpética: aciclovir 200 mg cada 4 h (omitiendo toma nocturna) durante 5 días, valaciclovir 500 mg cada 12 h durante 5 a 10 días, o famciclovir 250 mg cada 8 h durante 5 días.
Terapia supresiva: pueden usarse los fármacos anteriores a dosis medias o bajas durante 6 a 12 meses; por ejemplo, aciclovir 400 mg cada 12 h, valaciclovir 500 mg cada 24 h, o famciclovir 250 mg cada 12 h. Es importante ajustar dosis según función renal del paciente y estado inmunológico del mismo.
2) Varicela
La varicela es una enfermedad vírica cutánea generalizada, muy contagiosa, de curso benigno en la infancia. Causada por la primoinfección del virus varicela–zoster (VVZ) de la familia Herpesviridae. Este virus es muy similar en morfología y capacidad de replicación a los demás Alphaherpesvirus: posee un genoma ADN (doble hebra lineal y aproximadamente 125 kpb), nucleocápside icosaédrica, tegumento amorfo y manto externo.
Mayoritariamente afecta a la piel y a las células de las terminaciones nerviosas sensitivas, donde pueden observarse cuerpos de inclusión típicos en el núcleo celular que actúan como agentes de memoria de replicación viral, activándose ante un deterioro del estado inmunitario del huésped, con más frecuencia en la tercera edad y en inmunodeprimidos.
Patogenia e inmunidad
El contagio es principalmente por vía aérea, aunque también se puede producir por contacto directo con lesiones cutáneas activas desde un individuo con varicela o herpes zóster. La probabilidad de contagio desde pacientes con herpes zóster es mucho menor que desde pacientes varicelosos. El VVZ penetra por vía conjuntival u orofaríngea, replicándose en el epitelio respiratorio y las amígdalas, para alcanzar posteriormente los ganglios linfáticos locorregionales. Tras pasar a sangre, continúa su replicación en los leucocitos polimorfonucleares periféricos, que contribuyen a su diseminación hematógena y a la llegada del virus a piel, mucosas y aparato respiratorio.
La transmisión tiene lugar desde un día antes de la aparición de las lesiones hasta la curación final de las vesículas, siendo más contagiosa durante los dos días previos a la erupción e inmediatamente posterior a ésta. Desde el punto de vista histopatológico, las lesiones son iguales que las producidas por el herpes simple y el zóster, con degeneración de la epidermis y edema que da lugar a vesículas con carga viral y células inflamatorias, y acaban formando costras que suelen curar sin dejar cicatriz.
Manifestaciones clínicas
El periodo de incubación dura de 14 a 21 días, suele ser asintomático en niños, y puede cursar con fiebre y cefalea en los adultos durante 24-48 horas. El exantema se presenta inicialmente en cara y cuero cabelludo, extendiéndose rápidamente a tronco y desde ahí al resto del cuerpo, afectando a piel y mucosas. El periodo exantemático dura unos 5-10 días; inicialmente es maculo-papular, después evoluciona hacia lesiones vesiculares rodeadas de halo eritematoso y finalmente forma costra. Suelen coincidir lesiones en distinta fase evolutiva, dando lugar a un patrón en cielo estrellado, típico de la varicela. La presentación clínica es más severa, con posibles complicaciones (Tabla 1), en los casos secundarios dentro de un mismo hogar, en los adultos y en estados de inmunidad celular alterada.
El VVZ puede infectar durante el embarazo y periodo neonatal, aunque es poco frecuente. La varicela congénita se define cuando la madre adquiere la enfermedad durante el primer trimestre de la gestación y da como resultado alteraciones en el feto en hasta en un 20% de los casos. La varicela del neonato es más frecuente, dándose cuando ocurre el contagio desde una semana antes del parto hasta 5 días después del mismo.
Diagnóstico
El diagnóstico de la varicela es eminentemente clínico en pacientes sanos; la analítica general solo muestra alteraciones inespecíficas (leucopenia y linfocitosis). Se debe realizar un diagnóstico diferencial con herpes simple, herpes zóster diseminado, foliculitis generalizada, eccema herpético, papulosis linfomatoide, pitiriasis liquenoide, impétigo ampolloso y síndrome de Stevens-Johnson, entre otros.
La confirmación diagnóstica está limitada por su accesibilidad, pero consiste en el aislamiento de virus vivos (Test de Tzanck), pruebas serológicas (por ejemplo, ELISA, que presenta una sensibilidad del 98%) y técnicas de biología molecular para detección de ADN viral (PCR). En el caso de embarazadas expuestas a un contacto con VVZ hay que recordar que el hecho de haber pasado la infección o de estar vacunada es ya un marcador fiable de protección; de lo contrario, se realizará un test ELISA, sobre todo si la gestación está en las últimas 2-3 semanas.
Tratamiento
Dependerá de la edad del paciente y de la gravedad del cuadro, siendo imprescindible la adopción de medidas para evitar una sobreinfección de las lesiones. El uso de solucionesantisépticas y astringentes, y de antihistamínicos (si aparece prurito), son soluciones eficaces. Nunca se indicará ácido acetilsalicílico por su relación con el Síndrome de Reye. Es aconsejable aislar a los pacientes hasta la resolución de todas las lesiones, así como el uso de antibióticos tópicos si se presenta sobreinfección de las mismas.
En niños sanos no está indicado el tratamiento específico de forma rutinaria. En niños mayores de 12-13 años parece que el tratamiento con aciclovir oral durante 7 días, dentro de las primeras 48 horas, podría acortar el número de días con fiebre y el número máximo de lesiones. Por otra parte, en pacientes con neumonía varicelosa y gestantes en tercer trimestre es preceptivo el tratamiento con aciclovir oral; en inmunodeprimidos, es preferible la vía endovenosa.
Por lo general, el pronóstico es excelente salvo si existe la sospecha de que aparezcan complicaciones en las que habrá que tener en cuenta la necesidad de derivación a nivel hospitalario (Tabla 2).
Prevención
La pauta de vacunación estándar comprende dos dosis de vacuna con un mínimo intervalo de un mes (efectividad 92-95% para prevenir cualquier forma de varicela). El Comité Asesor de Vacunas de la Asociación Española de Pediatría recomienda la primera dosis de vacuna frente a varicela a los 12-15 meses y la segunda a los 2-3 años. Simultáneamente, se mantiene la recomendación de vacunación de rescate a los 12 años, así como de las personas con factores de riesgo y sus contactos.
3) Herpes zóster (HZ)
La infección por el herpes zóster (HZ) se manifiesta como una erupción aguda de distribución metamérica originada por la reactivación del virus varicela zoster, que permaneció en estado latente, acantonado en los ganglios sensitivos del paciente. La incidencia de HZ se estima en 2-3 casos por cada 1.000 habitantes y año. Se estima que el 50% de las personas que viven más de 80 años podrían padecer al menos un episodio de reactivación a lo largo de su vida (Tabla 3). El HZ es infrecuente en niños y adultos jóvenes, aunque se ha detectado mayor incidencia en aquellos que adquirieron la varicela intraútero o durante el primer año de vida.
La morbilidad suele quedar reducida al dolor que persiste tras la infección activa, conocido como neuralgia postherpética. La afectación oftálmica puede causar pérdida temporal o permanente de la agudeza visual, incluso ceguera. Entre las complicaciones, aunque infrecuentes, destacan la sobreinfección de las lesiones, o la afectación meníngea o visceral, que pueden provocar morbilidad adicional.
Fisiopatología
Durante la primoinfección por el VVZ, éste se integra en el ADN celular del huésped no permitiendo su eliminación por parte del sistema inmunitario. Se produce una replicación viral “latente”, de modo que cuando el sistema inmunitario falla, la replicación se vuelve eficaz, provocando la aparición de lesiones típicas de HZ. Parece ser que factores como la radiación, traumatismos, fármacos u otras infecciones o situaciones de estrés también pueden facilitar la reactivación vírica. Se piensa que el HZ aparece cuando los títulos de anticuerpos y la inmunidad celular descienden por debajo de ciertos niveles que prevendrían la invasión viral.
Clínica
El HZ se manifiesta en las regiones inervadas por una o más ganglios espinales craneales. Las metámeras más frecuentemente afectadas van desde C2 a L2, junto con los pares craneales V y VII.
Los pródromos suelen ser leves con astenia, clínica catarral, fiebre y cefalea. La primera manifestación suele ser la aparición de dolor, en ocasiones intenso, y parestesias en el trayecto del nervio infectado. A veces, el HZ presenta una exclusiva afectación neuropática, sin presencia de lesiones cutáneas (“herpes sine herpes”). La erupción característica aparece 2-3 días después de que el virus llegue a la piel, en forma de pequeñas vesículas que se hacen más numerosas en 3-5 días, para posteriormente romperse, ulcerarse y secarse hasta formar costras. Las lesiones suelen aparecer en el territorio correspondiente a un solo dermatoma (Tabla 4) y en un hemicuerpo, aunque en ocasiones excepcionales se observan de manera bilateral (herpes dúplex).
Formas Clínicas del herpes zóster:
Intercostal: es el más frecuente, suele afectar uno o dos pares de raíces y ganglios intercostales.
Braquial: suele afectar a metámeras C8 y D1.
Lumbar: poco habitual.
Cervical: lesiones en la región anterolateral del cuello.
Cervico occipital: afecta a las tres primeras raíces y ganglios cervicales.
Trigémino: es el más frecuente de los pares afectados, destacando la afectación de la rama oftálmica y de las nasociliares, lo cual podría llegar a presentar una afectación corneal, con aparición de lesiones irreversibles; son típicas las lesiones en punta de la nariz (signo de Hutchinson).
Zóster del geniculado: afecta al gusto y la sensibilidad lingual, con parálisis facial asociada (síndrome de Ramsay-Hunt)
Glosofaríngeo: cursa con glosodinia y dolor en base lingual, con aparición de lesiones en mitad del velo palatino.
Herpes diseminado: en pacientes con enfermedad de base grave, sobre todo en la enfermedad de Hodgkin y en SIDA; llega a presentar en casos severos afectación visceral.
La neuralgia posherpética (NPH) es la complicación más frecuente del VVZ y consiste en la persistencia de parestesias y dolor tras un mes del comienzo del zóster. Los pacientes suelen describir dolor quemante, urente, continuo, que empeora durante la noche y puede ser desencadenado por estímulos no dolorosos. Los factores de riesgo para llegar a desarrollarla son: edad mayor de 50 años, pródromos sensitivos e inmunodepresión (trasplantados y VIH).
Diagnóstico
Esencialmente clínico: se sospecha ante la aparición de lesiones típicas y la distribución metamérica y se refuerza con los antecedentes de varicela. Las pruebas de laboratorio restringirse a situaciones de presentación atípica o para establecer un diagnóstico diferencial en inmunodeprimidos. En este caso, la técnica de elección es la detección directa de antígeno viral de las lesiones y el cultivo viral.
Tratamiento
De manera similar ante la presencia de otras infecciones de presentación vesicular, se recomiendan soluciones antisépticas. En caso de sobreinfección, se utilizarán antibióticos tópicos (mupirocina o ácido fusídico). En caso de herpes oftálmico, se aplica aciclovir tópico cada 4 horas, hasta al menos 3 días tras la curación de las lesiones.
En lo referente al tratamiento sistémico, se pueden emplear aciclovir, valaciclovir y famciclovir; estos dos últimos, no autorizados en niños. El aciclovir tópico no está indicado y debiera restringirse su uso endovenoso exclusivamente en mayores de 50 años, inmunodeprimidos, con afectación ótica u oftálmica y siempre que no hayan transcurrido 72 h tras la aparición de las lesiones.
La NPH se trata con antidepresivos tricíclicos (por ejemplo, amitriptilina), opiáceos o anticomiciales.
Pronóstico
El HZ suele resolverse espontáneamente en 2-3 semanas y es rara su reaparición. La afectación de nervios motores puede provocar parálisis temporal o permanente, siendo esta una importante complicación, así como la afectación ocular que puede llegar a producir ceguera.
Virus del papiloma humano (VPH)
Los virus del papiloma son virus de ADN, de pequeño tamaño (unos 50-55 nm de diámetro), que contienen aproximadamente 7.900 pares de bases, incluidos en la familia de los Papovavirus, que se caracterizan por producir clínicamente lesiones tipo verruga. Las verrugas son lesiones exofíticas queratósicas y son las manifestaciones clínicas más frecuentes que aparecen como resultado de la infección por el VPH (Tabla 5).
Los papilomas pueden afectar a múltiples animales siendo específicos para cada especie. Se comportan causando mutaciones del ADN que provocan el desarrollo de tumores en humanos, monos, ciervos, caballos, bóvidos, perros, pájaros y conejos. Se han identificado más de 130 tipos, que son clasificados según semejanzas en su secuencia genética, que se correlaciona con las tres categorías usadas para describir las enfermedades por VPH: anogenital y/o mucosa, cutánea no genital y epidermodisplasia verruciforme.
Se estima que hasta el 10% de la población presentará en algún momento de su vida verrugas vulgares, llegando hasta el 33% de los niños de educación primaria y con un pico de incidencia en la adolescencia; esa frecuencia aumenta en personas inmunodeprimidas. Se trata de un virus que se transmite tanto por contacto directo como por fómites contaminados por personas infectadas. El periodo de incubación es largo, entre 2 y 12 meses. Se inactiva fácilmente con solución jabonosa, éter o alcohol.
Manifestaciones clínicas
Verruga vulgar o filiforme. Se asocia a los genotipos 1, 2, 4 y 7, los cuales crean lesiones hiperqueratósicas, sobre-elevadas, por lo habitual menores de 1 cm de diámetro, pero que pueden confluir. Suelen ser del color de la piel o blanquecinas, delimitadas por halo rosado. Son más frecuentes en el dorso de las manos (Figura 1) y en zonas periungueales, rodillas, región perioral y tobillos. Habitualmente involucionan espontáneamente en el plazo de uno a dos años.
Verruga plantar. Causada por el VPH 1, aparece como lesión hiperqueratósica no excrecente y dolorosa a la palpación, sobre todo al pellizcamiento lateral. Un signo que facilita su diagnóstico es la presencia de pequeños puntos negros en su interior, que corresponden a trombosis capilares. En ocasiones, pueden confluir formando verrugas en mosaicos.
Verrugas planas. Se originan por infección por el VPH 3. Son lesiones más pequeñas, no queratósicas, y suelen ser múltiples, especialmente frecuentes en niños; se localizan generalmente en cara y manos, con tendencia a la hiperpigmentación en verano.
Condilomas acuminados. Aparecen como consecuencia de infecciones por los subtipos VPH 6 y 11. Presentan un aspecto papilomatoso y consistencia blanda, de localización anogenital, y son debidos a una transmisión sexual. Su presencia en la edad puberal debe hacer sospechar de abusos sexuales, máxime si existen lesiones anales (Figura 2) o sobre el esfínter. Si están a más de 3 cm del esfínter anal, probablemente se deban a inoculación de sus padres o cuidadores cuando éstos presentan verrugas vulgares en los dedos o manos.
Condiloma gigante otumor de Buschke-Löwenstein. Es una lesión que se desarrolla sobre una verruga anogenital preexistente. Clínicamente, se manifiesta como placas infiltradas de aspecto tumoral mamelonado que llega a borrar las características anatómicas de la región donde se asienta; se comporta como un tumor localmente invasivo con poca capacidad para producir metástasis a distancia. Histológicamente, es similar a un carcinoma escamoso bien diferenciado.
VPH y cáncer. Los subtipos 16 y 18, principalmente, se relacionan con la inducción de displasias y neoplasias. Es ampliamente conocida su relación con el carcinoma de cérvix, aunque desempeñan también un papel favorecedor para la aparición de otras lesiones malignas, como es el caso del carcinoma escamoso cutáneo u otros tipos de neoplasia aún no clarificadas. Se consideran factores de riesgo para la aparición de cáncer: la multiplicidad de parejas sexuales, el haber padecido otras infecciones de transmisión sexual, una historia personal de existencia de verrugas genitales o el tener compañeros sexuales con neoplasia intraepitelial.
Diagnóstico
Como en el caso de otras infecciones anteriormente descritas, el diagnóstico es esencialmente clínico. En caso de duda, puede necesitarse una biopsia, que revelará una hiperplasia epidérmica con células vacuoladas e inclusiones intranucleares e intracitoplasmáticas. Para demostrar la presencia del virus, es necesario el empleo de microscopía electrónica o técnicas de hibridación del ADN y de inmunohistoquímica. Además, es importante realizar un diagnóstico diferencial de las lesiones producidas por el VPH (Tabla 6).
Tratamiento
No existe tratamiento específico para el VPH, de forma que los diferentes tratamientos disponibles van dirigidos a la destrucción física de las células infectadas por el virus (o facilitar la función del sistema inmunitario). El objetivo terapéutico será, pues, eliminar las lesiones cutáneas intentando respetar al máximo la piel sana, si bien es importante tener en cuenta que la mayor parte de las verrugas desaparecen espontáneamente en el transcurso de meses o años. En cualquier caso, ningún tratamiento es efectivo al 100%. Las personas inmunodeprimidas suelen responder peor al tratamiento, obteniéndose los mejores resultados en pacientes jóvenes con lesiones de corta evolución.
Para el tratamiento de verrugas vulgares se suelen emplear queratolíticos potentes –como el ácido salicílico y láctico al 16% en una base de colodión–, crioterapia (mediante spray de nitrógeno líquido), electrocoagulación o un láser de CO2. En el caso de verrugas planas, se recurre a agentes queratolíticos (ácido retinoico, salicílico o urea) o láser de CO2. De forma similar, las verrugas plantares suelen tratarse con queratoliticos potentes, 5-fluoracilo tópico (agente quimioterápico que destruye lesiones verrucosas) o con crioterapia mediante nitrógeno líquido; se deben evitar la cirugía y la electrocoagulación por el riesgo de aparición de cicatrices retráctiles dolorosas.
Por último, el tratamiento de los condilomas acuminados implica el uso de podofilino, una resina compuesta por citotóxicos cuyo componente más activo es la podofilotoxina. Es útil únicamente en lesiones localizadas en mucosa, por su dificultad para atravesar la capa córnea; se contraindicada durante el embarazo y no debe aplicarse en amplias zonas corporales por riesgo de toxicidad sistémica. Otra opción es el imiquimod al 5%, un fármaco inmunomodulador que elimina las lesiones por modificación de la respuesta inmunológica al inducir la síntesis local de citocinas.
Prevención
El empleo del preservativo reduce la posibilidad de contagio del VPH, aunque solo evita un 70% de los casos debido al contacto de las zonas genitales no cubiertas o a su uso inadecuado.
La citología mediante la técnica de Papanicolau es fundamental como técnica de cribado, contribuyendo de manera determinante a la reducción de la morbilidad y mortalidad por cáncer de cérvix en más de un 75% de personas en las poblaciones en las que se utiliza de forma sistemática y continuada, gracias a la detección de lesiones pre-neoplásicas.
Pero la base de la prevención de esta infección por VPH es la vacunación profiláctica, que evita eficazmente la infección persistente por los VPH y el desarrollo de las lesiones pre-neoplásicas que ocasiona: constituye una estrategia preventiva de primer orden frente a las neoplasias ano-genitales y, de forma específica, para el cáncer de cérvix. El desarrollo de las vacunas profilácticas para VPH se ha centrado en una proteína estructural de la envoltura externa del virus obtenida mediante técnicas de ingeniería genética (proteína L1). Esta proteína se autoensambla cuando se expresa en cultivos de células eucariotas y forma partículas similares al virus (virus-like particles o VLPs) que son capaces de inducir una respuesta inmunitaria de anticuerpos neutralizantes que permite prevenir la infección por VPH; al no contener genoma viral, las VLPs no pueden causar infección ni tienen potencialidad para causar lesiones neoplásicas. Pero se debe recordar que las vacunas son meramente profilácticas, ofreciendo la posibilidad de prevenir la infección inicial por el VPH frente a los genotipos incluidos en ellas, sin efecto terapéutico alguno sobre la infección ya establecida ni sobre las potenciales lesiones secundarias a la misma.
Dermatosis por poxvirus
Los poxvirus son virus ADN de tamaño entre 250 y 300 nm, de replicación citoplasmática, simetría compleja y sin envoltura. Las principales manifestaciones cutáneas producidas por infecciones por poxvirus son la viruela, el molluscum contagiosum, el nódulo de los ordeñadores y la Enfermedad de Orf.
Molluscum contagiosum
Se trata de un proceso vírico benigno, frecuente en niños, que puede afectar piel y mucosas, siendo adultos a menudo una enfermedad de transmisión sexual. Su agente etiológico es un poxvirus exclusivamente epidermotrópico, del genero Mollucipoxvirus, del que existen cuatro subtipos: el más frecuente es el tipo 1, mientras que el tipo 2 asociado a inmunodeprimidos. Su extensión geográfica es universal, la mayoría de los casos se producen por contagio de persona a persona y mediante pequeñas soluciones de continuidad de la piel, lo que podría explicar la mayor afectación en individuos atópicos (debido al rascado).
Los mecanismos de contagio son diferentes en niños y en adultos. Mientras que en niños siempre aparecen en epidemias localizadas en escuelas o zonas recreativas comunes, sobre todo piscinas (aunque también podría existir transmisión instrumental por fómites, pues el virus no se inactiva al desecarse), en adultos existe un pico entre los 15 y los 29 años en que aparecen lesiones en el área genital a través del contagio sexual. También es posible el contagio vertical produciéndose la contaminación en el canal del parto.
La célula diana del mollucipoxvirus es el queratinocito, en cuyo citoplasma ocurre la replicación viral; ésta genera unos característicos cuerpos de inclusión más evidentes en el estrato granuloso y córneo de la epidermis, dando lugar a la hiperproliferación epidérmica al doblarse el índice de división celular en la capa basal.
Con un periodo de incubación es de 2 a 7 semanas, en niños las lesiones –que pueden ser muy diseminadas– suelen afectar a tronco, axilas, cara, zona perianal y perigenital. Clínicamente, las lesiones son pápulas cupuliformes y umbilicadas de 3 mm a 3 cm de diámetro que aparecen sobre piel sin lesión previa. Pueden presentarse aisladas o agrupadas, y son lesiones asintomáticas, que por compresión lateral pueden expulsar material blanquecino en el que se encuentran multitud de partículas virales. Dichas lesiones suelen desaparecer a largo plazo (hasta 1 año), periodo durante el cual el paciente puede presentar multitud de moluscos, ya que por autoinoculación el proceso puede extenderse y las recidivas son muy frecuentes. Además, en pacientes inmunodeprimidos o con eccema crónico o atópico, pueden aparecer formas intensas y diseminadas.
El diagnóstico de esta infección es sencillo y eminentemente clínico; hay que realizar diagnóstico diferencial con verrugas vulgares, varicela, miliaria sudoral y liquen nitidus. En caso de dudas, se puede realizar biopsia: el estudio histopatológico se caracteriza por crecimiento de la epidermis hacia la dermis en forma de lóbulos cuyos queratinocitos tienen en el citoplasma grandes cuerpos de inclusión con alta carga viral.
El tratamiento debe ser individualizado en función del paciente, teniendo en cuenta que ninguna opción modifica el curso ni la recidiva de los moluscos. Los efectos estéticos o psicoterapeúticos del curetaje pueden ser útiles en ocasiones. Otras alternativas terapéuticas son: la crioterapia, la electrofulguración, el pellizcamiento con pinza, el uso de hidróxido de potasio o ácido retinoico. La administración de fármacos inmunomoduladores, como imiquimod, puede ayudar a resolver la infección viral. La cimetidina a altas dosis durante 3 meses ha demostrado eficacia en ensayos clínicos.
Nódulos de los ordeñadores
Se trata de una infección vírica cutánea producida por un poxvirus del mismo subgrupo (parapoxvirus) que el virus de la enfermedad de Orf. Este virus es endémico en el ganado vacuno, en el que produce lesiones vesiculosas en las ubres, transmitiéndose por contacto directo a las manos de los ganaderos. No se ha comprobado la transmisión directa entre humanos. Cada vez es menos habitual debido a la mejora de las condiciones laborales en el sector agropecuario.
Clínicamente, se trata de una lesión única nodular, purpúrea en la región central y eritematosa en la periferia, localizada en las manos y de un tamaño que oscila entre 1 y 5 cm. Asintomática o poco dolorosa, cursa sin sintomatología general. Estas lesiones pueden ser múltiples por auto-inoculación (no > 4 lesiones) pero evolucionan espontáneamente hacia la curación.
El diagnóstico es clínico, y debemos sospecharlo ante la presencia de antecedentes de contacto con ganado; la biopsia es una opción en caso de dudas. Además, se debe realizar diagnóstico diferencial con la enfermedad de Orf, ántrax, granuloma piógeno, tuberculosis cutánea y carbunco.
Enfermedad de Orf (dermatitis pustulosa contagiosa o ectima contagioso)
Producida por un poxvirus del subgrupo parapoxvirus, muy similar al nódulo de los ordeñadores, la fuente de contagio es el ganado lanar y caprino. Solo se da en la raza blanca y es más frecuente en primavera. Suele ser lesión única nodular de 1,5 cm de diámetro y de localización en región dorsal de los dedos de la mano (Figura 3). Es una infección autolimitada que se resuelve sin complicaciones de forma habitual en 3 o 4 semanas.
Dermatosis causadas por retrovirus humanos. VIH y coronavirus SARS-CoV-2
La apremiante actualidad nos obliga a incluir en esta revisión las lesiones cutáneas producidas por retrovirus.
La historia de la patología debido a la acción de los retrovirus humanos se inicia en la década de los 80 con el descubrimiento del virus de la leucemia de células T humano (HTLV–1), agente causante de la leucemia de células T del adulto. En 1981 también se describió la enfermedad por el virus VIH (virus de la inmunodeficiencia humana) como un síndrome de inmunodeficiencia adquirida caracterizado por la presencia de infecciones y neoplasias oportunistas, lesiones herpéticas ulcerosas crónicas y sarcoma de Kaposi. En 1983 los grupos de Mountaigner y Gallo aislaron los virus denomiandos LAV y HTLV–III y en 1985 pudieron demostrar que eran el mismo virus y el causante del sida (VIH). Este virus puede dar lugar a diversas manifestaciones cutáneas (Tabla 7).
Por su parte, los coronavirus constituyen un amplio grupo de virus que se encuadran taxonómicamente en la subfamilia Coronaviridae. Se trata de virus cuyo genoma está formado por
una única cadena de ARN con polaridad positiva y de aproximadamente 30.000 pares de bases, que presentan una capucha metilada en el extremo 5‘ y una cola poliadenilada en el extremo 3‘, que les aportan un gran parecido al ARN mensajero del hospedador; ello les permite adherirse a los ribosomas del huésped para su traducción. Por microscopía electrónica, los viriones se reconocen por una pequeña corona que presentan a su alrededor y que justifica su nombre.
Se tratan de virus zoonóticos, esto es, pueden transmitirse de animales a humanos, siendo los mamíferos reservorios u hospedadores intermediarios; destacan, entre ellos, los murciélagos, en los que se facilita la recombinación y los eventos mutagénicos favorecedores de una mayor diversidad genética vírica. En la infección de los mamíferos, colonizan fundamentalmente el tracto respiratorio superior y el tracto gastrointestinal. Los más conocidos por su patogenicidad hasta ahora eran el MERS-CoV, o coronavirus causante del Síndrome Respiratorio de Oriente Medio, y el SARS-CoV, responsable del Síndrome Respiratorio Agudo y Severo.
Según análisis genéticos, el nuevo coronavirus descrito a finales de 2019 pertenece al grupo de los betacoronavirus y guarda un estrecho parentesco con SARS-CoV, por lo que finalmente ha sido denominado SARS-CoV-2 por la OMS. Las principales manifestaciones de la enfermedad que provoca, la COVID-19, incluyen: fiebre, tos, disnea e infiltrados pulmonares bilaterales en la radiografía torácica. En muestras de autopsias realizadas al inicio de la pandemia en China se observó exclusivamente un infiltrado linfocitario perivascular inespecífico en dermis superficial, pero no se han descrito cambios específicos en la piel debido a la infección por SARS-CoV-2 ni se observó evidencia de éste en la piel.
Las manifestaciones cutáneas de la COVID-19 serían muy variadas e inespecíficas; además, podrían no tener relación con la gravedad del cuadro y resolverse de manera espontánea. Las formas de presentación cutánea que se han descrito hasta el momento son las siguientes (Figura 4):
Exantemática: eritematosa, petequial, morbiliforme. Puede ser generalizada o localizada, de predominio en tronco, poco o nada pruriginoso. Suele aparecer al inicio de los síntomas o días después, y es de resolución espontánea.
Erupción urticariforme: se ha descrito en hasta en un 1,4% de los pacientes con COVID-19, indistinguible de una urticaria aguda.
Erupción vesicular de tipo varicela.
Lesiones acroisquémicas o pseudoperniosis: asintomáticas inicialmente, posteriormente pueden provocar dolor. Aparecen en zonas distales (manos y pies), son lesiones de escasos milímetros y bien delimitadas, que suelen darse en cara lateral, dorso y punta de dedos, plantas y talones. Evolucionan durante unas 2 semanas, tornándose purpúricas y pudiendo aparecer, en su superficie, vesículas, necrosis y hasta gangrena seca (aunque esta última solo se ha descrito en pacientes graves hospitalizados).
Otras lesiones: livedos reticularis o púrpura petequial folicular; se investiga la asociación con síndromes inflamatorios multisistémicos que guardan estrecha relación en su manifestación con la enfermedad de Kawasaki.
Bibliografía
Albrecht MA, Levin MJ. Epidemiology and pathogenesis of varicella-zoster virus infection: Herpes zoster. Up To Date, Waltham, MA versión 19.3. 2011. Disponible en: https://www.uptodate.com/contents/epidemiology-clinical-manifestations-and-diagnosis-of-herpes-zoster.
Alcántara Muñoz P, Ortiz Díaz F, Maestro Saavedra FJ. Coronavirus y manifestaciones cutáneas. Actualización en Medicina de Familia. Disponible en https://amf-semfyc.com/web/article_ver.php?id=2650.
Asociación Española de Pediatría. Virus del Papiloma Humano. Manual de vacunas en línea de la AEP. Disponible en: https://vacunasaep.org/documentos/manual/cap-42 (acceso a junio de 2020).
Braue A, Ross G, Varigos G, Kelly H. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005; 22(4): 287-94. DOI: 10.1111/j.1525-1470.2005.22401.x.
Corey L, Mandell G, Benett J, Dolin R. Virus Herpes simplex. Enfermedades infecciosas Principios y Práctica. Buenos Aires. Editorial Panamericana. 2000; 1911-31.
Chen Y, Peng H, Wang L, Zhao Y, Zeng L, Gao H et al. Infants Born to Mothers With a New Coronavirus (COVID-19). Front Pediatr. 2020; 8: 104. DOI: 10.3389/fped.2020.00104.
Jiménez Alés R. Enfermedades víricas de la piel. Pediatr Integral. 2012; XVI(3): 222-34.
López Rocha A, Sabio Reyes F, Sánchez Camacho R. Guía de Buena Práctica Clínica en Infecciones víricas dermatológicas. OMC y Ministerio de Sanidad y Consumo. 2005. ISBN: 84-689-1940-3. Disponible en: https://www.cgcom.es/sites/default/files/guia_dermatologia.pdf.
Monteagudo B, Cabanillas M, Acevedo A, Pérez-Pérez L, Suárez-Amor O, Ginarte M. Infants Born to Mothers With a New Coronavirus (COVID-19). Front Pediatr. 2020; 8: 104. DOI: 10.3389/fped.2020.00104.
Peteiro García C, Gómez Vázquez M. Infecciones víricas de la piel. Clasificación. Formas Clínicas. Criterios Diagnósticos. Actitudes terapéuticas. Medicine. 2002; 4744-52.
El mieloma múltiple (MM) o mieloma de células plasmáticas es un tumor hematológico agresivo de células o linfocitos B, caracterizado por la proliferación en la médula ósea de un clon de células plasmáticas malignas (células mielomatosas) que desplazan a las células plasmáticas normales y, generalmente, producen y secretan una paraproteína monoclonal, detectable en suero o en orina como signo subyacente al daño orgánico; además, pueden formar tumores localizados, los plasmocitomas, de localización ósea mayoritaria, pero también extraósea. El MM representa aproximadamente el 1% de los casos de cáncer y, específicamente, el 10% de los tumores hematológicos a nivel mundial, con una incidencia de 4-6 casos por 100.000 habitantes/año en la población occidental. Se estima que en España hay cerca de 6.000 pacientes con MM, y la Sociedad Española de Oncología Médica ha calculado que en 2020 se diagnosticarán un total de casi 3.200 casos nuevos, con una afectación ligeramente superior de los varones (ratio 1,4:1). La mediana de edad al diagnóstico es de unos 65-67 años (se considera excepcional antes de los 40 años) y prácticamente todos los pacientes evolucionan desde estadios pre-malignos asintomáticos llamados gammapatía monoclonal de significado incierto o MM indolente.
De etiología multifactorial y no claramente elucidada, el MM tiene una fisiopatología compleja y su diagnóstico no es sencillo, pudiendo retrasarse varios meses al pasar en muchas ocasiones desapercibido en las consultas de atención primaria por la inespecificidad de sus manifestaciones: síntomas de tipo gripal, fracturas y dolor óseo, anemia, estreñimiento, etc. Aunque se considera como una patología aún incurable (algunos autores hablan de que está en vías de curación) y de difícil manejo, cabe destacar que el tratamiento ha logrado grandes progresos en las últimas dos décadas, duplicando la esperanza de vida de los pacientes hasta medianas de supervivencia de casi 5 años, gracias al mejor conocimiento de la patobiología celular, la mejora de estudios citogenéticos y técnicas diagnósticas y de estadiaje, los avances en el control de las comorbilidades y de los eventos adversos de los tratamientos, y la aparición de nuevos fármacos. Además de los “clásicos” agentes alquilantes y corticosteroides, se dispone ahora de una variedad de fármacos, como los inhibidores de proteasoma, agentes inmunomoduladores o anticuerpos monoclonales, cuyas combinaciones –junto al trasplante autólogo de médula en pacientes candidatos– en las fases de inducción, consolidación, mantenimiento y recaída han permitido abrir la posibilidad de individualizar el abordaje y cronificar la patología.
El presente artículo revisa ampliamente el conocimiento actual sobre la etiopatogenia, la epidemiología, las manifestaciones clínicas y el tratamiento del mieloma múltiple, centrando el foco en los frutos del avance de la investigación biomédica. Se pone en valor, finalmente, el importante papel asistencial que el profesional farmacéutico, desde sus distintos ámbitos de actuación, puede ejercer para con los pacientes con mieloma múltiple, en términos de educación sanitaria, detección precoz y optimización de la farmacoterapia.
Introducción
El mieloma múltiple (MM) o mieloma de células plasmáticas es un agresivo tumor hematológico maligno de células o linfocitos B, caracterizado por la proliferación en la médula ósea de un clon de células plasmáticas (procedentes de la maduración de células B) que, generalmente, producen y secretan una paraproteína monoclonal, detectable en suero o en orina (banda en forma de “pico” en el proteinograma electroforético) como signo subyacente al daño orgánico. Presenta algunas características similares a la leucemia y supone aproximadamente el 1% de los casos de cáncer y, específicamente, el 10% de los tumores hematológicos.
Grosso modo, las neoplasias hematológicas engloban a todos aquellos procesos de origen tumoral que afectan al tejido hematopoyético y al sistema linfoide. Se considera tejido hematopoyético a la médula ósea y todo su complejo sistema celular. Por su parte, el sistema linfoide integra a los ganglios, tejido linfoide de diferentes órganos (y bazo, fundamentalmente), y a todos los procesos que afectan a elementos celulares, como linfocitos B y T y células plasmáticas (Figura 1).
Una clasificación muy general de las neoplasias hematológicas, incluyendo su incidencia en el mundo occidental, se presenta en la Tabla 1. En conjunto, este tipo de tumores suponen algo más del 10% de los tumores en humanos, siendo más frecuentes en la edad avanzada (su incidencia se ve multiplicada hasta en 10 veces a partir de los 80 años de edad), sobre todo las leucemias agudas y gammapatías; la excepción la representan las leucemias linfoides agudas, que son la principal causa de cáncer infantil, y el linfoma de Hodgkin (LH), que ocurre en edades medias de la vida.
Al contrario que en los tumores sólidos, no existen registros exhaustivos de la incidencia y prevalencia de algunos de estos procesos. En unas ocasiones, porque acontecen en edad muy avanzada y, en otras, porque son procesos neoplásicos poco agresivos que conviven con otras enfermedades de base del paciente y no son comunicados, como es el caso de la leucemia linfática crónica en los estadios iniciales. De forma similar, los cuadros mixtos denominados síndromes mielodisplásicos no tienen siempre un carácter maligno y en sus primeras etapas pueden cursar solo con anemia u otras citopenias, por lo que pueden estar infradocumentados; no obstante, son en muchas ocasiones cuadros pre-leucémicos de base neoplásica.
El mieloma múltiple (MM) se enmarcaría dentro de las llamadas gammapatíasmonoclonales: un grupo heterogéneo de enfermedades caracterizadas por una producción anormal de inmunoglobulinas y la aparición de tumores de células plasmáticas. Dentro de ellas, otros procesos pueden considerarse pre-neoplásicos, como es el caso de la gammapatía monoclonal de significado incierto (GMSI). En cualquier caso, este trastorno asintomático, que también implica la proliferación de un clon celular de células plasmáticas y está presente hasta en más del 3% de la población mayor de 50 años, precisa de monitorización continua (no de tratamiento), dado que un 1% anual puede derivar en MM. En algunos pacientes, se identifica un estado premaligno intermedio asintomático más avanzado que se denomina como mieloma múltiple latente; la tasa de pacientes con este trastorno que progresan a MM es del 10% en los primeros 5 años, decreciendo posteriormente (3% los años siguientes y 1-2% anual a partir de los 10 años).
Desde un prisma histórico, si bien el MM ha estado presente en la sociedad durante miles de años, el primer caso bien documentado fue descrito en 1844 (Solly, 1844). Se trataba de una mujer de 39 años que desarrolló fatiga y dolor óseo derivado de múltiples facturas. En la autopsia posterior a su muerte, 4 años después del inicio de los síntomas, se halló que la médula ósea había sido reemplazada por una sustancia roja1 cuyas células eran muy similares a las encontradas en la autopsia de Thomas Alexander McBean, quizá el caso más conocido y previo al anterior.
Este señor era un comerciante británico que, a los 45 años de edad desarrolló fatiga y notaba que su «ropa de cama estaba rígida por su orina»; estando de vacaciones en septiembre de 1844, notó un crujido seguido de dolor severo en el pecho, que le impedía moverse. Tratado con una escayola de yeso y la aplicación de sanguijuelas como terapia de mantenimiento, el dolor desapareció, para volver a recurrir a los pocos meses; en esa situación, el uso de ventosas y flebotomía terapéutica no fueron eficaces. Tras prescribírsele acero y quinina, mejoró rápidamente, pero en otoño de 1945, el dolor intenso reapareció y falleció el primer día de 1946. En su autopsia, se vieron costillas frágiles y ligeramente fracturadas, encontrándose en sus huesos una «sustancia gelatiniforme de color rojo sangre y tacto untuoso». El examen histológico de la médula ósea reveló células redondas u ovales de tamaño variable (tenían entre la mitad y hasta el doble del tamaño de una célula sanguínea normal) y contenían 1-2 núcleos y nucleolo de color brillante.
El reputado patólogo inglés Henry Bence Jones estudió detalladamente la orina del paciente, que algunos de sus colegas habían definido como “orina que tiene una alta gravedad específica; cuando se hierve, se vuelve ligeramente opaca, y con la adición de ácido nítrico, se torna efervescente, de tono rojizo y bastante clara; cuando se enfría, toma consistencia y apariencia específicas, mientras que el calor la licua”. El Dr. Jones concluyó que la proteína presente en la orina era el «deutóxido hidratado de la albúmina». La presencia de esa proteína, que a la postre acabaría llevando el nombre de su descubridor, es uno de los signos característicos del MM. Por otro lado, el término «enfermedad de Kahler» con que en ocasiones se designa al MM, es resultado de un informe de un médico llamado Dr. Loos por el profesor Otto Kahler, de Praga. El paciente en cuestión tenía dolor óseo progresivo y proteinuria con las características típicas de la proteína de Bence Jones (concretamente, cadenas ligeras libres monoclonales); en la autopsia, se observó la presencia de células grandes y redondas compatibles con MM.
Con la descripción precisa de las células plasmáticas a finales del siglo XIX por distintos autores, entre los cuales Ramón y Cajal fue posiblemente el pionero, y la siguiente introducción en 1929 de la técnica de aspiración de médula ósea, el diagnóstico del MM progresó y mejoró sustancialmente. Por ejemplo, Rosenthal y Vogel (Rosenthal et al., 1938) informaban de que solo se habían reconocido 3 casos de MM en el Hospital Monte Sinaí de Nueva York desde 1916 hasta 1935, pero que se encontraron 13 casos en los siguientes 2 años y medio. Destacaron la presencia de fracturas patológicas, proteinuria de Bence Jones, anemia y enfermedad renal crónica. Sin embargo, no reconocieron las anormalidades de la velocidad de sedimentación o las proteínas sanguíneas. El posterior avance de la investigación biomédica permitió la identificación de los subtipos de cadenas ligeras de los anticuerpos, así como la diferenciación entre gammapatías monoclonales y policlonales; dicha diferenciación es relevante porque los pacientes con gammapatía monoclonal pueden tener o llegar a desarrollar un proceso neoplásico maligno mientras que la presencia de una gammapatía policlonal es indicativa de un proceso inflamatorio o reactivo (Kyle et al., 2008).
Etiopatogenia
Tal y como se ha sugerido previamente, las células plasmáticas malignas o cancerosas características del MM –células mielomatosas– crecen de forma descontrolada y desplazan a las células plasmáticas normales, se acumulan en la médula ósea y pueden llegar al torrente sanguíneo, provocando una alteración de la función fisiológica de la médula ósea, daños óseos y alteración de la función inmunitaria. Las células mielomatosas pueden también formar tumores localizados, los plasmocitomas (masas de mieloma), que pueden tener una localización tanto ósea como extraósea; justamente si coexisten varios plasmocitomas, con diversas localizaciones, es cuando se emplea apropiadamente el término mieloma múltiple.
Pero, sin duda, la característica más típica de la célula mielomatosa es la producción y liberación al torrente circulatorio de una inmunoglobulina monoclonal, denominada proteína M, también conocida como proteína mielomatosa, para-proteína o proteína en pico (esto último es debido a su característica determinación mediante electroforesis). Se trata de una inmunoglobulina producida como consecuencia de la aparición de una o más mutaciones en los genes responsables de la producción de inmunoglobulinas en la célula mielomatosa. Tiene una secuencia de aminoácidos y una estructura anormales en relación a las inmunoglobulinas fisiológicas, lo que provoca la adherencia entre las moléculas y con las estructuras celulares y de tejidos: células sanguíneas, pared de vasos sanguíneos u otros componentes de la sangre. Todo ello provoca la disminución del flujo sanguíneo, causando un síndrome de hiperviscosidad que determina la afectación multiorgánica.
El componente monoclonal es de tipo IgG en el 53% de los casos de MM, IgA en un 25% e IgD o IgE en menos del 3%. Otras variantes menos frecuentes son los MM no secretores (1% de los casos) o los MM biclonales, así como la presentación o evolución del MM a leucemia de células plasmáticas (> 20% de plasmocitos en sangre periférica).
Sin embargo, en un 20-30% de los casos de MM no se observa componente monoclonal detectable en suero, pero se produce una mayor cantidad de cadenas livianas –de tipo kappa o lambda– que las requeridas para combinar con las cadenas pesadas de las inmunoglobulinas, dando lugar a la proteína de Bence-Jones. Esta tiene un peso molecular de 22 kDa (kilodaltons) y es lo suficientemente pequeña como para ser excretada con la orina y producir un aumento del nivel de proteínas urinarias. Estas proteínas, igual que la inmunoglobulina completa, también pueden adherirse entre sí o con otros tejidos, dando lugar a amiloidosis (depósitos proteicos en cualquier tejido del organismo, como riñón, tejido nervioso o músculo cardiaco) o a la enfermedad de depósito de cadenas livianas: las cadenas livianas se depositan al azar, pero especialmente en los pequeños vasos del ojo o del riñón. Las proteínas monoclonales anormales también pueden provocar un amplio abanico de perturbaciones fisiológicas, al unirse a los factores de la coagulación –generando trastornos diversos de coagulabilidad– y a otras sustancias circulantes, con diversas consecuencias hormonales y metabólicas (Cuéllar, 2018a).
Por otro lado, cabe destacar el papel de la Beta 2 microglobulina (β2M): una pequeña proteína cuyos niveles se encuentran elevados en casos de mieloma en actividad, aunque un 10% de los pacientes no la producen. Precisamente, como se tratará más adelante, el estadiaje internacional del MM se basa en las concentraciones en suero de albúmina y de microglobulina β2.
A grandes rasgos, el crecimiento descontrolado de las células mielomatosas tiene importantes consecuencias, como la destrucción del esqueleto, la insuficiencia de la médula ósea, hipervolemia e hiperviscosidad sanguíneas, supresión de la producción de inmunoglobulinas normales e insuficiencia renal. Las células mielomatosas y el aumento del número y activación de los osteoclastos parecen ser las responsables de la destrucción ósea. El mecanismo que produce la activación de osteoclastos es complejo, pero se ha observado que participan diversas citocinas locales, tales como interleucinas (IL-1b, IL-6) y factor de necrosis tumoral alfa (TNFα), quimiocinas como las MIP-1 y las integrinas implicadas en el proceso de adhesión celular.
Entre las citocinas con implicación en la patobiología del MM, quizá la mejor caracterizada sea la IL-6, que es producida fundamentalmente por células madre de la médula ósea y macrófagos, y parece ser un importante mediador para el crecimiento, supervivencia y migración celular del mieloma, e incluso de la resistencia a la quimioterapia. Por un lado, actúa como un estímulo paracrino de las células plasmáticas, y, por otro, su secreción por el estroma de la médula ósea se ve estimulada por la adhesión de las células plasmáticas; se genera así un auténtico sistema de retroalimentación que potencia la tumorigénesis, con la IL-6 potenciando la supervivencia de las células plasmáticas e inhibiendo su apoptosis. El efecto acumulativo de este complejo ambiente bioquímico, con desequilibrio de fuerzas anti- y proapoptóticas dentro de las células de MM, favorece la expansión clonal desregulada y la proliferación tumoral.
Actualmente, se considera que los factores efectores finales que regulan la remodelación ósea forman parte de la superfamilia del Factor de Necrosis Tumoral (TNF) y de la de su receptor. Entre ellos puede citarse el RANKL (Ligando del Receptor del Activador del Factor Nuclear Kappa-B), cuya producción es máxima en las células indiferenciadas del estroma osteoclástico y se reduce a medida que madura el fenotipo osteoblástico. Estimula la diferenciación, supervivencia y fusión de las células precursoras de osteoclastos, activa los osteoclastos maduros y prolonga su vida útil. Como resultado permite la expansión de la masa osteoclástica activa capaz de formar sitios de resorción ósea.
Las células mielomatosas tienen, además, una serie de características estructurales y funcionales específicas que se describirán en el apartado de Tratamiento, por estar estrechamente relacionadas con los mecanismos de acción de algunas de las opciones terapéuticas disponibles.
A pesar de los eventos fisiopatológicos descritos hasta aquí, la etiología del MM, como para la práctica totalidad de tumores, se considera multifactorial. Si bien no se conocen con exactitud las causas directas, y aunque no se ha detectado un factor genético concreto que determine el desarrollo de la patología, suelen predominar eventos oncogenéticos primarios o secundarios2 que originan una proliferación descontrolada de un clon celular neoplásico. De igual modo, se ha asociado el MM con la exposición a radiaciones ionizantes, y también se ha propuesto un mayor riesgo si existen exposiciones ocupacionales –a sustancias químicas, como benceno, asbesto, pesticidas o pinturas y disolventes– relacionadas con la agricultura, refinerías, industrias del corcho, del metal, del plástico o de la madera, o si se ha trabajado como conductor de camiones.
Adicionalmente, se ha sugerido la posible relación del MM con algunas enfermedades autoinmunes, como la artritis reumatoide, o infecciones por algunos virus. El tratamiento previo con quimio/radioterapia por otra neoplasia o las situaciones de inmunodeficiencia (como ocurre en personas de edad avanzada) también podrían conllevar un mayor riesgo de padecer MM (Cuéllar, 2018b). Es más probable que el MM se desarrolle en individuos genéticamente propensos que se hayan expuesto a uno o más de los anteriores factores.
No obstante, las bases moleculares de la transformación inicial de las células plasmáticas normales a células mielomatosas, originariamente en el establecimiento de la GMSI, son inciertas. Parece que en las fases muy tempranas está implicada la desregulación de la familia de proteínas de ciclina D (ciclinas D1, D2 y D3), posiblemente mediante traslocaciones de los genes CCND1 (ciclina D1) y CCND3 (ciclina D3) con el gen IgH, amplificaciones específicas de genes de las ciclinas D, trisomías, o bien otros eventos citogenéticos aún no caracterizados. La sobreexpresión de las ciclinas D emerge, pues, como un evento necesario en la anormalidad temprana de las células plasmáticas, pero que por sí solo no explica la evolución hasta el establecimiento del MM (Brigle et al., 2017). En su desarrollo desde GMSI o MM latente, se han planteado como factores de riesgo la presencia de: un componente monoclonal distinto a IgG, mayores niveles de proteína monoclonal, cadenas ligeras libres anormales y ciertas alteraciones genéticas (Michels et al., 2017). En la siguiente Tabla 2, se recogen algunas de las alteraciones genéticas más comúnmente encontradas en pacientes con MM, así como el riesgo que comportan en el desarrollo y en el pronóstico de la patología.
En este sentido, diversos estudios que han empleado las modernas técnicas de secuenciación genética de próxima generación han demostrado la falta de una única mutación específica que guíe el desarrollo del MM, sino más bien al contrario: la presencia de varios subclones de células plasmáticas malignizadas por distintas mutaciones con cierto grado de solapamiento. Hay cierto consenso en esa heterogeneidad mutacional que se traduce en una diversidad clonal, si bien la asociación entre la evolución de los subclones con la progresión de la enfermedad aún no ha sido elucidada. Los genes más frecuentemente mutados en el MM corresponden a genes KRAS y NRAS (con una incidencia de aproximadamente el 25% cada una), implicados en las vías de señalización de MAP cinasas (MAPK) que regulan el crecimiento y supervivencia celular, seguidos de mutaciones en los genes que codifican para TP53 (hasta en el 15%), la exosoma endorribonucleasa DIS3 (11%), el gen de función incierta FAM46C (11%) o el gen BRAF (6-15%), también implicado en la vía de señalización de MAPK (Castaneda et al., 2019).
El establecimiento y la progresión del MM, como en otros tipos de cáncer, se ven favorecidos por su capacidad para suprimir y evadir el sistema inmunitario normal del huésped. De hecho, se ha descrito que la composición del sistema inmunitario presente en el microambiente de médula ósea de un paciente con MM es bastante diferente de la de un sujeto sano. Las células mielomatosas regulan una expansión de las poblaciones de células inmunosupresoras y la correspondiente disminución e inactivación de las células antitumorales, así como la sobreexpresión de los ligandos y receptores de muerte celular programada. Estos cambios inducen la tolerancia inmunitaria en un microambiente que permite la proliferación de las células tumorales.
En particular, se ha identificado un cambio en la composición de la población de linfocitos. Por un lado, se ha descrito un mayor número de células Treg CD4+ por acción de la IL-6 y el TGFβ (factor de crecimiento tumoral β), más abundantes de lo normal en el microambiente medular del MM: estas células tienen una función inmunosupresora (crean un ambiente inmunoprivilegiado para las células mielomatosas) y un mayor nivel de ellas en sangre se correlaciona con un menor tiempo hasta la progresión y una supervivencia global más corta; además, se cree que secretan factores que promueven el desarrollo de los osteoclastos y la subsecuente patología ósea. Por otro lado, se observa una disminución significativa del número y actividad de células T CD8+, las principales células con función citotóxica –junto a las células NK3 – implicadas en la respuesta antitumoral y vigilancia del tumor. Esas células CD8+ son funcionalmente diferentes respecto a las presentes en sujetos sanos, con mayor expresión de la proteína de muerte programada PD-1; cuando se unen a su ligando en las células mielomatosas (PD-1L), se activan vías de señalización que inhiben su función antitumoral natural. En base a este hecho, se estudian tratamientos de inmunoterapia que actúan sobre dicha vía de señalización PD-1/PD-1L.
Por último, también cabe destacar que, en el microambiente del MM, los elevados niveles de IL-6 y VEGF (factor de crecimiento endotelial vascular) incrementan la expansión y bloquean la diferenciación a células maduras de las células supresoras de línea mieloide, precursores con capacidad de diferenciarse a macrófagos, granulocitos y células dendríticas que normalmente están en bajos niveles en la médula ósea y tienen capacidad inmunosupresora (inhiben la proliferación de las células CD8+) (Brigle et al., 2017).
Epidemiología
El mieloma múltiple representa aproximadamente el 1% de todas las neoplasias y el 10% de las neoplasias hematológicas a nivel mundial, con una incidencia de 4-6 casos por 100.000 habitantes/año en la población occidental (en torno a 140.000 nuevos casos anuales en todo el mundo), que, aunque relativamente constante, puede oscilar ligeramente entre países. La mediana de edad al diagnóstico es de unos 65-67 años (el 85% de los pacientes tiene más de 50 años al diagnóstico y es excepcional antes de los 40 años) y, como ya se sugirió, prácticamente todos los pacientes diagnosticados evolucionan desde un estado pre-maligno asintomático llamado gammapatía monoclonal de significado incierto (GMSI), aunque algunos derivan de un estado intermedio, también pre-maligno y asintomático pero más avanzado, llamado MM indolente.
Se estima que su incidencia anual en Europa es de 4,5-6 casos por 100.000 habitantes, lo que se traduce en una cifra de en torno a 40.000 nuevos casos anuales; afecta tanto a mujeres como a hombres, siendo algo más común en estos últimos (ratio 1,4:1). En general, los países del sur de Europa tienen tasas de incidencia y mortalidad más bajas que los del norte; sin embargo, tanto en unos como en otros la mortalidad por esta patología está creciendo y se prevé que la incidencia aumente hasta más de 43.000 afectados anualmente para 2030.
En España, la Asociación Española de Afectados por Linfoma, Mieloma y Leucemia calcula que se diagnostican 4 nuevos casos de MM por cada 100.000 habitantes y año, representando una de las 5 neoplasias hematológicas más frecuentes. Algunos autores apuntan a que es la segunda o tercera neoplasia hematológica en incidencia tras los linfomas no Hodgkin y las leucemias. Si bien la incidencia anual es baja en menores de 65 años (1,5-2,5 casos por cada 100.000 habitantes), crece significativamente a partir de esa edad (hasta 25-30 casos nuevos por cada 100.000). Con esas tasas de incidencia, la Sociedad Española de Oncología Médica (SEOM, 2020) ha calculado que en 2020 se diagnosticarán en nuestro país un total de casi 3.200 casos de MM, con una afectación ligeramente superior de los varones (1.861 casos vs. 1.337 casos esperados en mujeres).
En Estados Unidos, algunos autores hablan de que el MM representa el 1,8% de todos los casos de cáncer y más del 17% de todos los tumores hematológicos. Con una tasa anualizada de incidencia estimada de 4,3 casos por cada 100.000 habitantes, se diagnostica más frecuentemente entre personas de 65 a 74 años, con una mediana de edad de 69 años. La Asociación Americana del Cáncer estima que en 2019 se diagnosticaron 32.110 casos nuevos y fallecieron 12.960 pacientes por MM (Kumar et al., 2019).
Se trata de una enfermedad incurable en la práctica totalidad de pacientes, que tiene una mortalidad elevada (4,1 muertes por cada 100.000 habitantes y año en Europa), representando un 2% de la mortalidad por cáncer y el 20% entre los tumores hematológicos. Por ello, la prevalencia no es muy alta: se estima que en España viven con la enfermedad unos 12.000 pacientes. No obstante, la supervivencia4 global se ha duplicado en los últimos 10 años con la introducción de nuevas terapias, y la mediana de supervivencia4 se ha prolongado hasta 45-60 meses desde el diagnóstico. Así, en España se estiman tasas de supervivencia a los 5 años que se sitúan en torno al 40-45% en varones y 48-51% en mujeres (EMA, 2016; SEOM, 2020). En todo caso, el MM tiene un elevado impacto sobre la salud de los pacientes, pero, también desde el punto de vista socio-económico, representa una carga importante para los sistemas de salud.
Aspectos clínicos
Manifestaciones
Al tratarse de una patología de células que circulan por la sangre, es posible que pueda saltar a cualquier órgano pudiendo generar plasmocitomas a distancia, si bien lo habitual es que afecte a la médula ósea y al hueso colindante, al menos inicialmente; son minoría –entre el 15-20%– los MM que debutan con masas localizadas, más comunes en casos avanzados o en recaída. Sin embargo, la enfermedad puede permanecer asintomática durante muchos años, en estadios premalignos. A grandes rasgos, el crecimiento descontrolado de las células mielomatosas tiene como una importante consecuencia la destrucción del esqueleto, la insuficiencia de la médula ósea, hipervolemia e hiperviscosidad sanguíneas, supresión de la producción de inmunoglobulinas normales e insuficiencia renal.
Existe un grupo de pacientes reducido (< 10%) con criterios biológicos de MM que debutan sin síntomas, en los que el diagnóstico se realiza por resultados alterados de análisis de sangre u orina (hipercalcemia, anemia, proteinuria), pero la mayoría de los pacientes con MM presentan daño orgánico con un grupo característico de síntomas designado con el acrónimo CRAB, siglas en inglés de: incremento de calcio, afectación renal, anemia y lesiones óseas. Las principales manifestaciones clínicas del MM se resumen a continuación (Alegre et al., 2017):
Cuadro constitucional con síntomas inespecíficos: astenia persistente y debilidad (en torno al 40% de pacientes), anorexia y pérdida de peso (25-30%).
Manifestaciones esqueléticas: es la sintomatología más frecuente (90%) y se debe a la destrucción de la masa ósea por la proliferación de las células plasmáticas y el aumento de la actividad de los osteoclastos; genera en los huesos llamadas “lesiones líticas”: zonas del hueso que pierden su contenido en calcio y se debilitan. El dolor óseo es el cuadro de presentación más común (70-80%), seguido de deformidades (60%) y fracturas patológicas (50%) por acumulación de lesiones. El dolor óseo, persistente o recurrente, suele localizarse sobre todo en la columna vertebral, costillas y caderas. Las fracturas se acompañan de un dolor intenso y de aparición brusca. A veces, el primer síntoma es un aplastamiento vertebral al levantar un peso no especialmente grande o una fractura después de un golpe banal.
Cuadro hematológico: anemia (70-90%), leucopenia y trombopenia. Menos frecuente es la diátesis hemorrágica por trombopenia o interacción del componente M con los factores de la coagulación; se debe a que las células mielomatosas invaden la médula ósea e impiden la normal producción de otras células sanguíneas.
Asociado a este cuadro hematológico, la inhibición de las funciones inmunitarias normales (producción deficitaria de anticuerpos normales, disfunción de los linfocitos T y activación anómala de la función monocito/macrófago) determina una mayor predisposición a infecciones, siendo los pacientes con MM particularmente susceptibles a infecciones virales y a infecciones por bacterias encapsuladas como el neumococo.
Manifestaciones renales: entre el 30 y el 50% de los pacientes presentan a lo largo de su evolución insuficiencia renal (IR) que suele ser plurietiológica. Un 20% debuta con IR aguda, en general reversible con el tratamiento.
Manifestaciones neurológicas: son frecuentes las neuralgias y radiculalgias por afectación vertebral. Más excepcionales son los cuadros de polineuropatía5 y el de encefalopatía o coma por hipergammaglobulinemia. La progresión en el conducto raquídeo de las masas mielomatosas puede originar un cuadro de compresión medular o desplazamiento de nervios procedentes del tallo cerebral, que precisa tratamiento urgente debido al riesgo de paraplejia secundaria.
Hipercalcemia: con cuadro característico de deshidratación, estreñimiento, somnolencia y hasta estupor; aparece en análisis de sangre en hasta el 15-30% de los pacientes en el diagnóstico de MM. Tanto la hipercalcemia como la insuficiencia renal requieren de tratamiento inmediato, aunque el diagnóstico de MM no esté aún instaurado.
Hiperuricemia: por elevada replicación celular, pudiendo originar litiasis renal por uratos.
Síndrome de hiperviscosidad por hiperproteinemia (sobre todo en MM con predominio de componente IgM), que determina un mayor riesgo de hemorragias, alteraciones visuales, respiratorias, etc.
Diagnóstico
El diagnóstico de mieloma múltiple requiere de la identificación de células plasmáticas clonales en la médula ósea o de plasmocitomas óseos o extramedulares en una biopsia. En muchas ocasiones el MM puede tardar en diagnosticarse, fundamentalmente porque con frecuencia los síntomas iniciales (dolor de espalda, cansancio, etc.) son poco alarmantes o fácilmente atribuibles a otras causas. No obstante, la frecuencia con la que actualmente nos hacemos análisis de rutina –sanguíneos o de orina– ha facilitado que cada vez sea más habitual diagnosticar un MM cuando todavía es asintomático, durante los controles de una GMSI detectada casualmente.
Los criterios específicos para el diagnóstico de un mieloma activo (Mikhael et al., 2019) han sido recientemente actualizados por el Grupo Internacional de Trabajo sobre Mieloma (IMWG, por sus siglas en inglés International Myeloma Working Group) e incluyen la presencia de células plasmáticas clonales más los signos CRAB o al menos uno de los tres nuevos biomarcadores (Tabla 3). Estos nuevos criterios, más estrictos, pretenden definir una población de pacientes con MM que son sintomáticos o pronto desarrollarán síntomas, por lo que requieren tratamiento urgente. Con su aplicación, muchos pacientes que previamente han sido diagnosticados con MM latente serán ahora definidos –más apropiadamente– como enfermedad activa y que requiere terapia; el objetivo es facilitar una detección y un inicio del tratamiento más tempranos que permitan mejorar la supervivencia y minimizar (o incluso evitar con futuras terapias) la actividad del MM.
Resulta importante llevar a cabo un diagnóstico diferencial para distinguir el MM de los ya citados estadios premalignos. Clásicamente se ha considerado que el MM latente viene definido por la presencia de: niveles de proteínas monoclonales en suero (IgG o IgA) ≥ 3 g/dl o de proteína monoclonal en orina ≥ 500 mg/24 h y/o niveles de células plasmáticas clonales en médula ósea entre 10% y 60%, pero sin eventos definitorios de MM (signos CRAB) o amiloidosis. La GMSI viene definida normalmente por una infiltración de células mielomatosas clonales < 10% en médula ósea y menor componente monoclonal en suero (< 3 g/dl de IgG o IgA o IgM), también sin daño orgánico y ausencia de signos CRAB atribuibles a la proliferación de células plasmáticas. Un subtipo específico, la GMSI de cadenas ligeras, está, además, determinada por un ratio anormal de cadenas κ/cadenas λ (< 0,26 o >1,65), un mayor nivel de cadenas ligeras alteradas (del tipo mayoritario), ausencia de cadenas pesadas de inmunoglobulinas en ensayos de inmunofijación y niveles de proteína monoclonal en orina < 500 mg/24 h.
Ante la aparición de alguno de los signos o síntomas ya citados, se debe realizar una detallada historia clínica del paciente, acompañándola con un examen físico que, en principio, puede ayudar a identificar otras causas de esas manifestaciones, si bien suele tener hallazgos normales en muchos pacientes con MM. Cuando existe la sospecha clínica, el diagnóstico de MM es relativamente sencillo. Deben realizarse dos pruebas de laboratorio esenciales que suelen ser suficientes para confirmar el diagnóstico: un análisis completo de proteínas en sangre y en orina que permita caracterizar correctamente el componente monoclonal (en < 95% de los casos de MM se observan proteínas monoclonales anómalas, que facilitará también el seguimiento en la respuesta al tratamiento) y un mielograma o aspirado de médula ósea6 (biopsia) para confirmar y cuantificar la presencia de células plasmáticas anormales en la médula ósea. También en el análisis de sangre se realizará un recuento completo de células sanguíneas, la determinación de los niveles de calcio, de albúmina y de creatinina.
En todo caso, es muy útil completar el diagnóstico con otras pruebas que permitan determinar el pronóstico y dirigir el tipo de tratamiento. A fin de determinar adecuadamente el número y localización de las lesiones óseas, se debe realizar un estudio radiológico de todo el esqueleto. La tomografía computarizada (TC) permite ubicar las lesiones líticas, siendo superior en sensibilidad a la radiografía simple. Por su parte, las imágenes obtenidas por resonancia magnética permiten evaluar mejor la columna vertebral, siendo especialmente interesante ante la sospecha de compresión medular o plasmocitomas junto a las vértebras. Por último, la tomografía por emisión de positrones (PET-TC) puede detectar lesiones en otras localizaciones, aunque no siempre es necesario hacer todas estas pruebas (Michels et al., 2017).
Pronóstico
La determinación del pronóstico en un paciente con MM suele tener en cuenta el estadio del tumor, determinado a partir de los criterios revisados del sistema internacional ISS de estadificación, que se basa mayoritariamente en las concentraciones en suero de albúmina y de microglobulina β2, y determina 3 niveles (Palumbo et al., 2015):
Estadio I: niveles de β2-microglobulina sérica < 3,5 mg/l y de albúmina > 3,5 g/dl; también niveles normales de lactato deshidrogenasa (LDH) y ausencia de anormalidades en el análisis por FISH. Se asocia a una mediana de supervivencia de más de 5 años (62 meses).
Estadio II: niveles de β2-microglobulina < 3,5 mg/l y de albúmina < 3,5 g/dl, o bien β2-microglobulina de 3,5 a 5,5 mg/l; en general, cualquier paciente que no cumple criterios de estadio I ni del III. La mediana de supervivencia se reduce a menos de 4 años (44 meses).
Estadio III: se caracteriza por niveles séricos de β2-microglobulina > 5,5 mg/l o niveles elevados de LDH. Mediana de supervivencia: 29 meses.
Clásicamente se han considerado algunos como índices principales de mal pronóstico: alta carga tumoral (estadio III del ISS, que suele relacionarse con mieloma sintomático: ECOG > 1, anemia, hemoglobina < 12 g/dl, hipercalcemia, alto componente IgM y lesiones osteolíticas múltiples), la presencia de insuficiencia renal (creatinina sérica > 2 mg/dl) irreversible tras el tratamiento, niveles de albúmina < 3 g/dl, niveles de β2-microglobulina sérica > 3 mg/l y de proteína C-reactiva > 0,4 mg/dl. La identificación de plasmocitomas al diagnóstico se asocia también con un pronóstico desfavorable, de enfermedad es más agresiva e invasiva. La edad avanzada, las comorbilidades, la mayor puntuación de la escala ECOG y la refractariedad al tratamiento son otros factores no dependientes de la enfermedad que implican mal pronóstico.
En la actualidad, el peso pronóstico más relevante lo determina el perfil citogenético, cuya determinación al diagnóstico es imprescindible y que define claramente 3 grupos pronósticos (estándar, intermedio y alto riesgo). Los hallazgos genéticos de peor pronóstico suelen realizarse por citogenética convencional: deleción del cromosoma 13, hipodiploidía o cariotipos complejos; y por la técnica de hibridación fluorescente in situ (FISH, por sus siglas en inglés): t(4;14), t(14;16), 17p (pérdida de la actividad supresora de tumores de TP53, que se relaciona con un pronóstico particularmente pobre) y amplificación 1q21. Todas, excepto esta última, también se consideran como criterios del estadio III del MM.
El principal problema es que el estadiaje del MM tiene realmente poca utilidad, pues lo que verdaderamente importa es la eficacia del tratamiento y, en particular, la respuesta a la primera línea tiene gran valor pronóstico. Así, la respuesta positiva y la negatividad de la enfermedad mínima residual tras un primer tratamiento –estimada por métodos moleculares de próxima generación o por citometría de flujo– tiene un pronóstico favorable, independientemente de que el paciente hubiera sido clasificado como estadio I, II o III. Por el contrario, aquel que no responde a un primer tratamiento o recae muy pronto tras una respuesta rápida inicial (un 10-15% de pacientes), tendrá un peor pronóstico, con supervivencias que se acortan hasta los 2 años en los casos más graves. Algunos autores apuntan a que alrededor del 20% de los pacientes con MM responden mal a cualquier tipo de tratamiento, mostrando el peor pronóstico. Además, otros pueden responder bien a un primer tratamiento, pero no responder a una segunda línea tras una recaída.
En resumen, el pronóstico del MM es ampliamente variable y las pruebas diagnósticas no permiten determinarlo con exactitud, ni en tiempo ni en calidad de vida ni de respuesta a tratamientos. Se considera, grosso modo, como una enfermedad aún incurable, en que la mayoría de pacientes sufrirán recaídas tras una respuesta al tratamiento inicial –desaparición de síntomas o mejora notable tras las primeras semanas de tratamiento– y necesitarán volver a tratarse, probablemente en varias ocasiones. Aunque sin riesgo vital a corto-medio plazo, el pronóstico del MM recidivante o recurrente a largo plazo es incierto, y depende de la frecuencia y agresividad de las recaídas: incluso con un pronóstico inicial similar, pacientes con recaídas poco agresivas y buena tolerancia a los tratamientos pueden tener buena calidad de vida durante años, mientras que otros con recaídas agresivas o mala tolerancia a fármacos, pueden tener manifestaciones clínicas que la deterioren notablemente. Solo una pequeña proporción de pacientes (≈10%) tendrá una excelente respuesta al tratamiento de primera línea y es posible que no recaigan nunca, considerándose como “funcionalmente curados”, aunque nunca se puede asegurar que el MM no vaya a reaparecer, incluso tras 10-15 años libres de enfermedad.
Tratamiento
Ante el diagnóstico de MM, la decisión terapéutica depende básicamente de si se trata de un paciente asintomático quiescente o bien de un paciente sintomático con clínica de MM; en general, los pacientes con MM latente7 o GMSI no suelen tratarse, y la opción preferente es la monitorización clínica, con controles periódicos cada 3-6 meses y medidas preventivas. El otro factor más relevante para decidir y planificar la terapia, aunque no el único, es la edad del paciente, debiendo diferenciar a los pacientes > 65-70 años de los de menor edad, pues el segundo grupo recibirá normalmente un tratamiento más intensivo, incluyendo la valoración del trasplante autólogo de progenitores hematopoyéticos.
Sea cual fuere la opción elegida, el objetivo debe ser individualizar el tratamiento para conseguir mantener el control de la enfermedad (sin recaídas, o minimizando la intensidad de éstas) y preservar la mayor calidad de vida de los pacientes durante el máximo tiempo posible. Se debe subrayar que la complejidad y heterogeneidad de la enfermedad aún dificulta en gran medida el desarrollo de una medicina personalizada; el desarrollo de técnicas genómicas de alto rendimiento, el mejor conocimiento de la biología del mieloma y la identificación de nuevas dianas permitirán el desarrollo de terapias cada vez más eficaces.
Fármacos anti-mieloma
En las últimas décadas –especialmente en los últimos 15-20 años– se ha asistido a grandes avances en el tratamiento del MM, con prolongación significativa de la esperanza de vida al diagnóstico: la tasa de supervivencia relativa a 5 años aumentó desde el 30% en la década de los 80 del pasado siglo a prácticamente el 50% en la segunda década del siglo XXI. Esta mejora se ha debido fundamentalmente a la introducción de nuevos fármacos, como el inhibidor de proteasomas bortezomib o agentes inmunomoduladores como lenalidomida, aprobados para el tratamiento de pacientes con MM hace ya más de 10 años. Pero otros fármacos también han contribuido notablemente al progreso terapéutico, como se verá a continuación; se recogen en la Tabla 4, y las estructuras químicas de la mayoría de ellos, en la Figura 2.
Desde la óptica del orden cronológico, en el siglo XX se dejaron atrás los remedios aplicados para el tratamiento de los primeros casos de MM descritos en la bibliografía científica, tales como preparados a base de ruibarbo, infusión de piel de naranja, flebotomía, la aplicación de sanguijuelas como terapia de mantenimiento o la quinina. Los primeros fármacos que se emplearon en los años 60 eran básicamente citostáticos tradicionales, de los que se usaban en todo tipo de cánceres. Así, Blokhin y colaboradores (Blokhin et al., 1958) describieron los primeros beneficios en esta enfermedad para melfalán (conocido inicialmente como sarcolisina): un antineoplásico del grupo de las mostazas nitrogenadas alquilantes que actúa como agente electrófilo, reacciona con átomos nucleofílicos de las bases nucleicas y forma puentes inter e intracatenarios en la doble hélice del ADN nuclear (sobre todo en la fase S del ciclo), interrumpiendo los procesos de transcripción y replicación del material genético celular. Trabajos subsiguientes en esa misma década de 1960 llegaron a constatar una tasa de respuesta de hasta el 78% con pacientes recién diagnosticados de MM. Quizá este fármaco continúa siendo el mejor agente único para el tratamiento del MM, y se recomienda como quimioterapia de acondicionamiento previa a la realización de la infusión de células madre en un trasplante autólogo de médula ósea.
—El mieloma múltiple representa un 1% de todas las neoplasias y el 10% de las neoplasias hematológicas a nivel mundial—
De forma paralela, los corticosteroides comenzaron a ser evaluados en diversos estudios, que demostraron que la prednisona como agente único aportaba una tasa de respuesta objetiva del 44%. Desde entonces, se estableció el régimen clásico de melfalán más prednisona (régimen MP), que exhibió –en un ensayo clínico con 183 pacientes– eficacia para prolongar la supervivencia en más de 6 meses en comparación con el uso de melfalán solo (Alexanian et al., 1969). El protocolo MP ha mostrado capacidad de inducir una respuesta objetiva en más de la mitad de los pacientes, manifestada con un 50% de mejora en los niveles de proteína M, en los recuentos sanguíneos y en otros resultados bioquímicos, además de la mejoría de varios síntomas de la enfermedad, como el dolor óseo y la fatiga. Más tarde se plantearon modificaciones de este régimen dual, con sustitución del melfalán por otras mostazas nitrogenadas alquilantes, como ciclofosfamida, con una actividad antimielomatosa similar, o bendamustina. Los resultados de esta última opción son equiparables a los obtenidos con melfalán; incluso, como régimen único, la tasa de respuestas completas es sustancialmente mayor con bendamustina y el porcentaje de pacientes supervivientes a los 5 años también, aunque en ambos casos moderado (29% vs. 19%).
La radioterapia comenzó por entonces a utilizarse con el mismo objetivo de suprimir las células mielomatosas. Actualmente, la radioterapia local representa una opción que puede ser muy efectiva en pacientes con importante destrucción ósea, intenso dolor y/o compresión nerviosa o de la médula espinal; su mayor inconveniente es que daña de forma permanente las células progenitoras normales de la médula ósea en el área tratada.
Más adelante, se introdujo la combinación de carmustina, melfalán, ciclofosfamida y prednisona y, ya en la década de 1970, se estableció la asociación de carmustina, ciclofosfamida, melfalán, vincristina y prednisona (bautizado como protocolo M-2), de la que se llegaron a reportar tasas de respuesta objetiva de hasta el 87% en un ensayo con 73 pacientes (Case et al., 1977). Sin embargo, un amplio meta-análisis (MTCG, 1998) con datos de casi 5.000 pacientes tratados demostró que, aunque esas combinaciones de quimioterapia aportaban mayores tasas de respuesta que el régimen MP (60% vs. 53%), no había diferencias significativas en duración de la respuesta o supervivencia global, por lo que este último se mantuvo como el estándar de tratamiento de inicio durante décadas (aún hoy se usa con cierta frecuencia). Las combinaciones más complejas se dejarían para una segunda línea en aquellos pacientes que no alcanzaran una respuesta satisfactoria.
Por otra parte, en los años 80 se inició y desarrolló la técnica del trasplante de células de médula ósea, primero alogénico y, después, autólogo, la opción que perduraría en la historia terapéutica del MM. El primer caso reportado de trasplante autólogo de progenitores hematopoyéticos (TAPH) de médula ósea (McElwain et al., 1983) se realizó en un paciente con leucemia de células plasmáticas, al cual se le administró melfalán (140 mg/m2) y, tras una recaída a los 16 meses, se le administró de nuevo dicho fármaco seguido de una infusión intravenosa de extracto de su propia médula ósea obtenida durante la fase de remisión. Hacia el final de la década, se desarrollaron los programas de tratamiento intenso usando trasplante autólogo, la conocida como “terapia total” (Barlogie et al., 1987), que jugaron un rol esencial en el establecimiento de la terapia de altas dosis de quimioterapia y de rescate con progenitores hematopoyéticos como estándar frente al MM en pacientes de nuevo diagnóstico y que están en buenas condiciones de salud general. Esta opción demostró mejorar la tasa de respuesta y la expectativa de vida de los pacientes, alcanzándose tasas de remisión completa de entre un 25% y un 75% (hay que tener en cuenta que con el resto de opciones que comentaremos, la mayoría de respuestas son parciales). No obstante, el 90% de los pacientes recaen, con tiempos medios de recaída de 18 a 24 meses; la expectativa global de vida en estos pacientes es de aproximadamente 4 a 5 años.
De manera interesante, fue a finales de los 90 cuando empezaron a aparecer toda una serie de moléculas antineoplásicas dirigidas más específicamente al mieloma y con menos efectos indeseables. Si bien los corticoides y agentes alquilantes siguen formando parte indispensable del tratamiento del MM, desde finales del pasado siglo, la incorporación de esos nuevos fármacos cambió de forma muy importante el pronóstico de la enfermedad, en parte porque exhibieron actividad en casos resistentes a los fármacos de primera generación. En general, ninguno de ellos se emplea aisladamente, y se acepta que un tratamiento más intenso y prolongado con una combinación de fármacos se traduce en una eficacia mayor (menor riesgo de resistencias y control más rápido de síntomas) y una mayor duración de la respuesta. No obstante, la intensidad y duración del tratamiento deben modularse para evitar un exceso de toxicidad: la individualización del tratamiento dependerá de la capacidad del paciente para tolerar tratamientos más o menos intensivos, siendo la edad un factor muy relevante para ello, aunque no el único a considerar.
Diferentes líneas de investigación facilitaron la incorporación de fármacos como bortezomib: un inhibidor del proteasoma 26S, el cual resulta ser una herramienta celular fundamental en el control del ciclo vital celular y de la apoptosis: degrada las proteínas ubiquitinadas y es esencial en la regulación del recambio proteico para mantener la homeostasis celular. La inhibición del proteasoma 26S evita la proteólisis dirigida y afecta a múltiples cascadas de señalización intracelulares, originando en última instancia la muerte de la célula neoplásica.
Bortezomib fue autorizado en Europa en 2004, inicialmente para el tratamiento de los pacientes con MM que habían recibido previamente al menos dos líneas de tratamientos y que presentaban progresión de la enfermedad demostrada con el último de estos tratamientos. Los datos clínicos disponibles indicaban unas tasas de respuesta del orden del 35% para respuestas completas y parciales, con periodos libres de progresión de la enfermedad de 9 a 13 meses en pacientes respondedores, muy superiores a los descritos hasta entonces en la bibliografía para pacientes con MM refractario o recidivante (alrededor de 3 meses). Posteriormente, fue eficazmente combinado con una administración intravenosa de doxorubicina liposomal, en un estudio clínico aleatorizado que demostró por primera vez la eficacia antimielomatosa de las antraciclinas (Orlowski et al., 2007). Futuros estudios permitieron la aprobación de bortezomib en combinaciones de primera línea en pacientes naïve no candidatos a trasplante de progenitores hematopoyéticos (con melfalán y prednisona) o candidatos a trasplante (con dexametasona y talidomida), y es uno de los fármacos más usados en la actualidad frente al MM (Scott et al., 2016).
El carfilzomib, autorizado en España en 2017, es un tetrapéptido con un grupo epoxicetona que representa una evolución dentro del grupo de los inhibidores del proteasoma: se une al extremo N terminal de los sitios activos del proteasoma 20S, el núcleo proteolítico del proteasoma 26S, y muestra poca o ninguna actividad frente a otros tipos de proteasas. Su efecto irreversible y su mayor selectividad sobre la función enzimática de tipo quimotripsina del proteasoma respecto a su antecesor bortezomib se traduce en un efecto antitumoral más potente y eficaz que el de éste (con mayor supervivencia libre de progresión y reducción de la tasa de progresión tumoral), pero también algo más tóxico. Está autorizado en combinación con lenalidomida y dexametasona o dexametasona sola para el tratamiento de adultos con MM que han recibido como mínimo un tratamiento previo. En esas combinaciones, carfilzomib aumentó las tasas de respuesta global hasta el 77-87% (vs. 67% con lenalidomida+dexametasona o 63% con bortezomib+dexametasona); en la combinación triple prolongó significativamente la SLP (supervivencia libre de progresión) y la SG (supervivencia global) hasta medianas de 26 meses y 48 meses, respectivamente (vs. 18 y 40 meses con lenalidomida+dexametasona).
El último fármaco de este grupo que ha sido autorizado en España –aún no comercializado– es el ixazomib, un inhibidor selectivo y reversible del proteasoma, que concretamente se une e inhibe la actividad tipo quimiotripsina de la subunidad β5 del proteasoma 20S; a diferencia de los dos fármacos anteriores (de administración parenteral), es activo por vía oral. De forma similar a carfilzomib, está autorizado, en combinación con lenalidomida y dexametasona, para el tratamiento de pacientes adultos que hayan recibido ≥ 1 terapia previa. Respecto al uso del doblete de lenalidomida y dexametasona, mejora significativamente la tasa de respuesta global (79% vs. 72%) y reduce el riesgo de progresión en un 26%, con medianas de SLP y de duración de la respuesta de casi 21 meses (vs. 15 meses).
Entre los fármacos inmunomoduladores activos por vía oral que también comenzaron a usarse a finales del siglo pasado, destacó inicialmente la talidomida; algunos estudios preliminares mostraron que es capaz de producir una significativa respuesta en pacientes con MM, con tasas de respuesta del orden del 25% en pacientes con MM recidivante/refractario, en su mayoría tras un doble trasplante. El problema que plantea este tratamiento es el de la toxicidad. De hecho, la talidomida se conocía desde varias décadas antes y había sido retirada (de su indicación como sedante y frente a las náuseas matutinas de las embarazadas) por su desgraciada implicación en numerosos y graves casos de dismorfogénesis fetal, calculándose que entre 1956 y 1962 más de 10.000 niños en todo el mundo nacieron con importantes deformidades, fundamentalmente focomelia.
Sin embargo, sus interesantes propiedades inmunomoduladoras atrajeron el interés de los científicos y algunos años después de su retirada se verificó su potencial terapéutico en múltiples patologías de tipo dermatológico, infeccioso y autoinmune. Asimismo, se comprobó su potencial antiangiogénico, siendo ensayada en diversos modelos de cáncer. Los resultados iniciales confirmaron que era eficaz frente al MM en todas las fases de la enfermedad, aportando tasas de respuesta en enfermedad en recaída de en torno al 50% combinada con corticoides, y del 65% si se combinaba con ciclofosfamida. Después, se han desarrollado varios regímenes de quimioterapia que incluían talidomida, estando a día de hoy autorizada –pero no comercializada– en España en combinación con melfalán y prednisona, con indicación de 1ª línea en pacientes mayores de 65 años o no aptos para recibir altas dosis de quimioterapia.
A pesar de haber sido autorizada en el pasado, incluso como medicamento huérfano, la toxicidad –sobre todo, teratogenicidad– de talidomida siempre ha sido el gran factor limitante de su uso. De ahí la necesidad de fármacos que mantuvieran sus propiedades, pero limitando su notable perfil toxicológico. Por ello se desarrollaron derivados como la lenalidomida, que no solo mantenían las propiedades de la talidomida sobre biomarcadores implicados en diversas patologías, sino que incluso las ampliaban, mejorando los resultados clínicos en MM, con un perfil toxicológico algo más benigno. Fue autorizado en 2007 en España tras demostrar tasas de respuesta interesantes en ensayos aleatorizados de fase 2: del 17% en monoterapia en pacientes en recaída o refractariedad, y de hasta el 91% en combinación con dexametasona en pacientes de nuevo diagnóstico. En subsiguientes ensayos pivotales de fase 3, la combinación con dexametasona se ha mostrado notablemente superior al uso de dexametasona sola tras recaídas (tras al menos 1 tratamiento previo). Además de para esa situación, actualmente se encuentra comercializada y es ampliamente empleada en monoterapia para el tratamiento de mantenimiento de adultos con MM de nuevo diagnóstico que se han sometido a un trasplante autólogo de células madre; también se indica –en varias combinaciones (con corticoides, brotezomib y/o melfalán)– para pacientes naïve no candidatos a trasplante.
La pomalidomida, autorizado en 2013, es un análogo de lenalidomida (de hecho, se trata de su oxoderivado) y comparte mecanismo de acción con ésta: se une directamente a la proteína cereblon, que forma parte de un complejo de ubiquitinación ligasa E3 (incluye la proteína reparadora 1 –DDB1– del ADN, culina 4 –CUL4– y regulador de culinas-1), y es capaz de inhibir la auto-ubiquitinación de cereblon dentro del complejo. Unidas a dicha proteína en células hematopoyéticas, ambos fármacos incorporan a las proteínas sustrato Aiolos e Ikaros, factores de transcripción linfoides, dando lugar a su ubiquitinación y su posterior degradación, lo que produce efectos directos citotóxicos e inmunomoduladores. Pomalidomida se emplea junto con dexametasona para el tratamiento de pacientes adultos con MM múltiple resistente al tratamiento o recidivante que hayan recibido al menos 2 tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad tras el último tratamiento. Y también asociada a bortezomib y dexametasona en adultos con MM en recaída tras ≥ 1 tratamiento previo, incluyendo lenalidomida. Con un perfil toxicológico similar o incluso algo más favorable, presenta una eficacia antimielomatosa mayor que sus antecesores (talidomida y lenalidomida).
En cualquier caso, un aspecto confuso del tratamiento del mieloma fue el descubrimiento de que una disminución de los niveles de la proteína mielomatosa en el suero y/o en la orina no se traslada necesariamente a una remisión clínica o a un aumento de la supervivencia. Dado que ningún tratamiento actual erradica todas las células mielomatosas, las características de aquellas residuales tras la quimioterapia inicial son de particular importancia. En este sentido, unas pocas células agresivas residuales pueden causar más problemas que una gran cantidad de células inactivas. Como se ha comentado previamente, el trasfondo genético del mieloma múltiple es complejo, lo que da como resultado una biología heterogénea que se traduce en una dificultad para desarrollar terapias dirigidas a las vías de señalización. Pero la expresión específica de varias moléculas de superficie en las células mielomatosas hace que sean potencialmente buenos objetivos para la inmunoquimioterapia.
Una de las dianas que ha despertado mayor interés en la terapéutica del MM es la molécula CD38, una glicoproteína transmembrana que es intensamente expresada por las células mielomatosas, aunque, en menor medida, también se expresa en diversos tipos de células linfoides y mieloides, e incluso en tejido no hematopoyéticos. La CD38 ejerce múltiples funciones, incluyendo actividad enzimática, regulación de la adhesión celular mediada por receptores y transducción de señales bioquímicas. La actividad enzimática implica la conversión de factores coenzimáticos, como NAD y NADP, en diversos sustratos requeridos para la regulación de la señalización intracelular del Ca2+. La CD38 también interviene en la adhesión al endotelio vascular, donde se acopla a la CD31 –su ligando natural presente en la superficie de las células endoteliales– y juega un papel relevante en la migración de los linfocitos. Asimismo, la interacción CD38/CD31 parece ser determinante en la señalización transmembrana, caracterizada por la movilización del Ca2+ y la secreción de diversas citocinas: en concreto, da lugar a la activación de linfocitos T, que inducen la secreción de IL-6 e IL-10, factores estimulantes de colonias de granulocitos y macrófagos (GM-CSF) e interferón gamma (IFN-γ).
El daratumumab es un anticuerpo monoclonal humano que se une específicamente a la molécula CD38 e impide sus acciones biológicas, activando diversos mecanismos citotóxicos e inmunitarios que acaban con la lisis o la apoptosis de las células que lo expresan, probablemente a través de citotoxicidad dependiente del complemento, citotoxicidad mediada por anticuerpos y fagocitosis celular dependiente de anticuerpos. Reduce el número de células cancerígenas, pero también se ven afectadas células T y B reguladoras normales y las células citotóxicas NK8 , aunque ello no impide que el recuento de linfocitos T en sangre periférica y médula ósea aumente significativamente, lo cual sugiere efectos inmunomoduladores que podrían contribuir a la respuesta clínica (Cuéllar, 2018a).
Fue autorizado en 2016 y, desde entonces, ha exhibido eficacia significativa en diversas situaciones clínicas y se han ido ampliando sus indicaciones autorizadas en MM, en combinación con otros fármacos: a) con lenalidomida y dexametasona o con bortezomib, melfalán y prednisona para tratar pacientes adultos de nuevo diagnóstico que no son candidatos a un trasplante autólogo de progenitores hematopoyéticos; b) con bortezomib, talidomida y dexametasona para el tratamiento de pacientes adultos de nuevo diagnóstico candidatos a trasplante; c) con lenalidomida y dexametasona, o bortezomib y dexametasona, para el tratamiento de pacientes adultos pre-tratados con ≥ 1 tratamiento previo. Incluso se ha autorizado en monoterapia para el tratamiento de pacientes adultos con MM en recaída y refractario al tratamiento, que hayan recibido previamente un inhibidor del proteasoma y un agente inmunomodulador y cuya enfermedad haya progresado.
Por otro lado, SLAMF7 (Signaling Lymphocyte Activation Molecule Family, SLAMF7), también conocido como CS1, CRACC o CD319 , es un miembro de la Familia de Moléculas de Activación de la Señalización de Linfocitos, que se sobre-expresa en células de MM, aunque también en células NK, linfocitos T CD8+, linfocitos B y células dendríticas maduras; no obstante, no se encuentra en tejidos sanos, tejidos tumorales primarios o líneas celulares de otros cánceres, hematológicos o no. CS1 es un receptor homofílico, y la interacción CS1-CS1 conduce a la activación de la citotoxicidad natural de las células NK. Parece haber una correlación entre CS1 soluble en el suero del paciente y el estadio de la enfermedad, lo cual indica que puede ser un biomarcador útil para la progresión de la enfermedad (EMA, 2016).
Elotuzumab es un anticuerpo monoclonal dirigido específicamente al SLAMF7 (CS1) que presenta un doble mecanismo de acción: por un lado, activa directamente a las células NK a través tanto de SLAMF7 como de los receptores Fc, potenciando de esta manera la actividad anti-mieloma y, por otro, al unirse al SLAMF7 de la célula mielomatosa, facilita su interacción con las células NK para mediar su destrucción, fundamentalmente a través de la citotoxicidad celular dependiente de anticuerpos. Elotuzumab ha sido el último principio activo nuevo comercializado en España para el tratamiento del MM, concretamente, en combinación con lenalidomida y dexametasona, para el tratamiento de adultos que han recibido ≥ 1 un tratamiento previo. Respecto al uso del doblete de lenalidomida+dexametasona solo, ha demostrado que reduce un 32% el riesgo de progresión y aumenta la mediana de SLP y SG en más de 4 meses (18,5 vs. 14,3 meses de SLP y 43,7 vs. 39,6 meses de SG), aumentando también la tasa de respuesta global (79% vs. 66%).
En paralelo a estos dos primeros fármacos de inmunoterapia, se aprobó en Europa panobinostat, que aún no está comercializado en España. Se trata de un inhibidor potente de histona deacetilasa (HDAC) por vía oral. La HDAC es una enzima que cataliza la eliminación de los grupos acetilo de los residuos de lisina de histonas y de algunas otras proteínas, por lo que su inhibición provoca una mayor acetilación de las histonas: una alteración epigenética que resulta en una relajación de la cromatina, y que activa la transcripción. Se ha descrito que este fármaco provoca la acumulación de histonas acetiladas y de otras proteínas, y promueve la expresión incrementada del gen supresor del tumor p21CDKNIA (inhibidor de la quinasa dependiente de ciclina 1/p21), mediador clave en el arresto de G1 y en la diferenciación; así, produce la detención del ciclo celular y/o apoptosis de algunas células transformadas, con una mayor citotoxicidad en células tumorales que en células normales. Panobinostat está indicado, en combinación con bortezomib y dexametasona, para el tratamiento de adultos con MM refractario y/o en recaída pre-tratados con ≥ 2 líneas previas con bortezomib y un agente inmunomodulador. Demostró una eficacia notable en un ensayo clínico que comparó la combinación autorizada frente a bortezomib+dexametasona solos: la triple terapia redujo a la mitad el riesgo de progresión del MM, con un incremento significativo de la mediana de SLP (12,5 meses vs. 4,7 meses) y de la tasa de respuesta global (59% vs. 39%).
Estrategias terapéuticas
En resumidas cuentas, la primera decisión de tratamiento ante un diagnóstico de MM se basa en la posibilidad de realizar o no un trasplante autólogo de progenitores hematopoyéticos (TAPH) de médula ósea. Conviene recordar que el trasplante alogénico (de un donante) no se recomienda de rutina en MM, pero sí podría considerarse en pacientes jóvenes de alto riesgo –según criterios revisados de estadificación ISS– en primera recaída o en el contexto de un ensayo clínico.
Pacientes candidatos a trasplante
Clásicamente, se ha considerado como candidatos a tratamiento intensivo y trasplante autólogo a aquellos pacientes más jóvenes (< 65-70 años) con un estado de salud general adecuado, si bien la guía de práctica clínica de la Sociedad Americana de Oncología Clínica –ASCO– (Mikhael et al., 2019) especifica que ni la edad ni la funcionalidad renal deben ser los únicos criterios tomados en cuenta a la hora de decidir la susceptibilidad de un paciente al TAPH.
En quienes se decida la conveniencia del trasplante, previamente se deberá administrar un régimen quimioterapéutico de inducción. Aún hay dudas sobre cuál es el régimen y el número de ciclos óptimo antes de la recolección de las células madre de médula ósea, pero hay cierto consenso en que se deben administrar al menos 3-4 (y hasta 6) ciclos de una terapia de inducción que incluya: un agente inmunomodulador, un inhibidor de proteasoma y un corticosteroide. Entre los regímenes de triple terapia preferidos, se encuentran el esquema BLD (bortezomib, lenalidomida y dexametasona9) o bien otros regímenes más recientes recomendados, como el CLD (carfilzomib, lenalidomida y dexametasona), especialmente en pacientes más jóvenes, o la combinación de ixazomib, lenalidomida y dexametasona (Kumar et al., 2019). Con estos esquemas se ha incrementado la tasa de respuestas globales y completas.
Otras combinaciones se consideran útiles en determinados tipos de pacientes o en ciertas circunstancias. Por ejemplo, si la lenalidomida no está disponible como terapia inicial o el paciente tiene insuficiencia renal aguda, puede considerarse su sustitución por talidomida o por ciclofosfamida en la combinación con bortezomib y dezametasona. Además, algunos pacientes pueden beneficiarse de posponer el TAPH después de la recogida de células hasta la primera recaída con el régimen de inducción, dependiendo de la profundidad de la respuesta, el riesgo y las preferencias de los pacientes.
Normalmente, fármacos asociados con toxicidad sobre las células madre de médula ósea, como melfalán o tratamientos prolongados con inmunomoduladores, deberían ser evitados en la fase de inducción. Sin embargo, una vez ya se han extraído las células progenitoras (se recomienda una extracción suficiente para más de un trasplante), melfalán a altas dosis es el régimen de acondicionamiento recomendado previamente a la realización del TAPH; no se ha demostrado superioridad de regímenes que combinan dicho fármaco a altas dosis con radioterapia de cuerpo entero o con otros agentes quimioterapéuticos (por ejemplo, busulfano, ciclofosfamida o bortezomib). Tampoco se ha establecido cuál es el nivel mínimo de respuesta requerido para proceder con el TAPH en pacientes que reciben regímenes de inducción, por lo que a priori todos deben proceder con el trasplante (infusión intravenosa de las células madre del propio paciente que se habían extraído y congelado previamente).
Tras el acondicionamiento y la realización del trasplante, no se recomienda de forma rutinaria la administración de una terapia de consolidación, que sí podría ser valorada en el contexto de un ensayo clínico o en pacientes que no quieren recibir tratamiento de mantenimiento. Por el contrario, un tratamiento oral de mantenimiento con lenalidomida (10-15 mg/día) sí se recomienda de forma general, empezando aproximadamente entre 3 y 4 meses después del trasplante y manteniéndolo hasta progresión. Un mantenimiento durante mínimo de 2 años se asocia con mejoras de la supervivencia (mediana de la SLP de hasta casi 53 meses vs. 23,5 meses con placebo) y con una reducción del riesgo de muerte en torno al 25% en comparación con placebo o ausencia de tratamiento (Gay et al., 2018).
No se dispone de evidencias suficientes para plantear modificaciones a esa terapia de mantenimiento en base a la profundidad de la respuesta o el estado de enfermedad mínima residual. Los pacientes intolerantes o en quienes se contraindique la lenalidomida, pueden beneficiarse de un mantenimiento con bortezomib cada 2 semanas, especialmente pacientes de riesgo intermedio-alto; también se está estudiando la eficacia de ixazomib en este contexto. Del mismo modo, para pacientes de alto riesgo (como aquellos con deleción 17p13) se puede valorar el mantenimiento con un inhibidor de proteasoma con o sin lenalidomida.
Se recomienda que se evalúe la profundidad de la respuesta en base a los criterios del IMWG10 tras cada ciclo del tratamiento de inducción, así como tras un máximo de 3 meses cuando se alcance la máxima respuesta o en la fase de mantenimiento. Para ello, la tomografía computarizada (TC) de dosis baja y cuerpo entero es el método preferido para la monitorización de la respuesta, pudiendo emplearse como alternativas la tomografía por emisión de positrones o la resonancia magnética.
—La mayoría de pacientes presentan los síntomas CRAB: aumento de la calcemia, afectación renal, anemia y lesiones óseas (sobre todo, dolor)—
Pacientes no candidatos a trasplante
Para aquellos casos que se consideren no elegibles para la realización de un TAPH, generalmente aquellos de edad más avanzada (> 65-70 años) o de peor estado general, el régimen terapéutico debe ser individualizado en base a una decisión compartida entre médico y paciente, que debe valorar múltiples factores, entre ellos, el estadio de la enfermedad, las anormalidades citogenéticas, edad, comorbilidades, estado funcional (incluyendo funcionalidad renal), fragilidad y preferencias del paciente.
Los regímenes terapéuticos deben incluir al menos uno de los “nuevos fármacos” (inhibidores de proteasoma o agentes inmunomoduladores) y, si es posible, un corticosteroide. Hace unos años, el esquema inicial más usado era el desarrollado por el grupo español de mieloma (GEM), denominado esquema VISTA, que combina bortezomib, melfalán y prednisona. A día de hoy, el esquema triplete recomendado de inicio es la administración conjunta de bortezomib, lenalidomida y dexametasona durante 8-12 ciclos. Alternativamente, también se considera como opción de primera línea la administración de un novedoso régimen cuádruple de daratumumab, bortezomib, melfalán y prednisona11.
En ausencia de estudios clínicos directamente comparativos, una reciente revisión sistemática y meta-análisis (Cao et al., 2019), que ha evaluado los datos de 23 artículos referentes a 10.401 pacientes naïve no candidatos a trasplante, ha concluido que los regímenes múltiples de daratumumab, dexametasona y lenalidomida (HR: 0,57; IC95% 0,43-0,73), daratumumab más bortezomib, melfalán y prednisona (HR: 0,59; IC95% 0,36-0,91), y la combinación de bortezomib con lenalidomida y dexametasona (HR: 0,72; IC95% 0,56-0,90) muestran una eficacia significativamente superior en términos de supervivencia libre de progresión en comparación con la asociación de lenalidomida y dexametasona; de forma interesante, ese beneficio se extiende más allá de los 75 años de edad de los pacientes. El triplete de bortezomib más dexametasona y lenalidomida ha sido el único que por ahora ha demostrado, además, un beneficio clínico para la supervivencia global en comparación con lenalidomida más dexametasona, con medianas de supervivencia de 75 meses vs. 64 meses (HR: 0,72; IC95% 0,53-0,96; p= 0,025).
Con el objetivo de alcanzar la máxima profundidad de respuesta (el mismo que en pacientes candidatos a trasplante), se acepta que el tratamiento debe administrarse de forma continua hasta progresión o intolerancia, estrategia que se ha mostrado superior respecto a la administración por un tiempo “corto” definido (por ejemplo, 18 meses). En todo caso, la decisión sobre la duración del tratamiento también debe involucrar al paciente, con consideración de sus preferencias, calidad de vida, toxicidad y costes de tratamiento.
Si bien los regímenes de 3 o 4 fármacos mejoran la respuesta y el control de la enfermedad en comparación con los regímenes de doblete disponibles (lenalidomida+dexametasona o bortexomib+dexametasona), también pueden aumentar la toxicidad del tratamiento, por ejemplo, respecto a la incidencia de neutropenia o neumonía (Facon et al., 2019) (ver Tabla 5). Por ello, la dosis inicial debe individualizarse en base a las características del paciente, para ajustarla después según la respuesta inicial y la tolerabilidad. Con independencia del régimen de primera línea, en estos pacientes no se recomienda un tratamiento de mantenimiento con lenalidomida.
Mieloma múltiple en recaída
Indudablemente, el principal problema del tratamiento del MM son las recaídas: casi todos los pacientes, a pesar de tener una buena respuesta a un primer tratamiento, recaen eventualmente tras una media de 1-3 años desde la primera remisión. En pacientes que presenten una respuesta (completa o parcial) muy duradera al primer tratamiento, la respuesta al segundo y sucesivos tratamientos suele ser también buena; en esos casos, podría considerarse el MM como una enfermedad crónica. Pronóstico muy diferente es el de aquellos pacientes que recaen muy pronto o de forma muy brusca y requieren rápidamente otro tratamiento, pues las recaídas favorecen la acumulación de secuelas (especialmente la insuficiencia renal y las lesiones óseas) y toxicidad, afectan a la calidad de vida y frecuentemente reducen las opciones de tratamiento siguientes. Así pues, cobra especial relevancia el anticiparse y evitar las secuelas de la enfermedad y los problemas relacionados con la toxicidad de los tratamientos realizando un seguimiento estricto de los pacientes con MM desde su diagnóstico.
La elección de la pauta terapéutica tras una recaída –o refractariedad12 a un primer tratamiento– es complicada, y se ve afectada por múltiples factores: el tiempo hasta la recaída, la respuesta a la terapia anterior (que podría repetirse si los resultados fueron muy buenos, debiéndose evitar fármacos que produjeron alta toxicidad), la agresividad de la recaída y
ciertas características del paciente: riesgo citogenético o comorbilidades (por ejemplo, neuropatía o insuficiencia renal), entre otras.
Una vez se confirma una recaída clínica del MM, los pacientes deben realizarse un tratamiento inmediato. Aquellos que son candidatos a trasplante TAPH deben ser considerados para dicho procedimiento si no se les ha practicado antes, o para un segundo TAPH si han tenido una remisión de duración excelente tras un primer trasplante (mediana de SLP ≥ 18 meses). No obstante, la práctica más común es el empleo secuencial de esquemas terapéuticos de rescate. La guía de práctica clínica de ASCO recomienda, ante una primera recaída, la administración de una triple terapia que incluya en combinación un inhibidor de proteasoma, un agente inmunomodulador y un corticosteroide, especialmente en pacientes con buen estado general, pues se han mostrado más eficaces que regímenes de doblete a base de uno de los “nuevos fármacos” con un corticosteroide. En cualquier caso, dado que la toxicidad suele ser mayor con un régimen de triplete, se debe considerar el uso de dobletes en pacientes que han experimentado toxicidad relevante con líneas de tratamiento previas.
Aunque sí que hay consenso sobre la conveniencia de mantener el tratamiento hasta progresión, con independencia del riesgo del paciente, la selección del mejor triplete (o cómo secuenciar tripletes con dobletes) no está aún del todo claro, en ausencia de comparaciones directas entre las alternativas disponibles. Como en el caso de pacientes de nuevo diagnóstico, los regímenes de bortezomib más lenalidomida más dexametasona, la combinación de bortezomib con ciclofosfamida y dexametasona y de bortezomib con talidomida y dexametasona son eficaces en el MM en recaída. También se han mostrado eficaces 3 combinaciones a base de daratumumab (con lenalidomida y dexametasona, con bortezomib y dexametasona, y con pomalidomida y dexametasona).
Otras opciones eficaces en el escenario de MM en recaída o refractario incluyen el uso de los fármacos de más reciente autorización: a) regímenes con ixazomib o elotuzumab (ambos combinados con lenalidomida y dexametasona); b) combinaciones a base de pomalidomida (por ejemplo, carfilzomib, pomalidomida y dexametasona); c) en casos de recaídas agresivas, se puede valorar el uso de regímenes que contienen doxorubicina; d) panobinostat; o e) regímenes que contengan bendamustina (como bendamustina+lenalidomida+dexametasona o bendamustina+bortezomib+dexametasona). Incluso el fármaco venetoclax, un inhibidor potente y selectivo de la proteína antiapoptótica BCL-2 indicado en leucemia linfocítica crónica, parece ejercer una actividad interesante en monoterapia en pacientes con un cierto subtipo de MM, aquél con la mutación t(11;14) (Rajkumar, 2019). En los casos multitratados, por la complejidad del abordaje, siempre es conveniente considerar la opción de ensayos clínicos con agentes en desarrollo en monoterapia o combinación.
—En general, se prefieren los esquemas terapéuticos triples: un agente inmunomodulador, un inhibidor de proteasoma y un corticosteroide—
Una revisión sistemática y meta-análisis de 17 ensayos clínicos aleatorizados y controlados de fase 3 (van Beurden-Tan et al., 2017) evaluó la evidencia disponible de hasta 18 opciones de tratamiento en MM en recaída y/o refractario. Los autores identificaron la combinación de daratumumab con lenalidomida y dexametasona como el tratamiento más eficaz y parece emerger como la opción preferente. Fue el régimen que aportó un mayor beneficio en términos de razón de riesgos o hazard ratio de SLP (HR= 0,13; IC95% 0,09-0,19), siendo el mejor régimen en el 99% de las simulaciones realizadas. Dicha combinación redujo el riesgo de progresión del mieloma o muerte en un 87% en comparación con dexametasona en monoterapia, en un 81% frente a bortezomib+dexametasona, y en un 63% frente a lenalidomida+dexametasona.
Otros meta-análisis posteriores más amplios, como el realizado por Maiese y colaboradores (Maiese et al., 2018), también concluyen que los regímenes a base de daratumumab (daratumumab con lenalidomida y dexametasona o daratumumab con bortezomib y dexametasona) son los que tienen una probabilidad más alta de aportar el mayor beneficio clínico en términos de supervivencia sin progresión tanto en pacientes que han recibido al menos 1 línea de tratamiento como en pacientes que han recibido solo 1 tratamiento previo.
En definitiva, como resumen de todo lo anterior, el MM es uno de los cánceres en los que se han aprobado más agentes farmacológicos con mecanismo singular y se siguen desarrollando nuevos esquemas de inmunoterapia, así como estrategias de mantenimiento preventivo una vez alcanzada la respuesta completa. Las recomendaciones generales sobre decisiones terapéuticas en función del tipo de paciente se reflejan en la Figura 3. Como en otras neoplasias, el elevado coste de las nuevas combinaciones (Tabla 6) obliga a un uso racional de las mismas, seleccionando los casos con mayor beneficio probable, de mayor relevancia –si cabe– en países sin sistemas de salud públicos.
Tratamiento de soporte
En líneas generales, además de los fármacos anti-mieloma específicos, los pacientes van a requerir otras terapias que se basan en dos pilares básicos (Alegre et al., 2017; GCEGM, 2017):
Medidas de soporte general:
Prevención de la nefropatía: hidratación abundante (con fluidos intravenosos si es necesario, para mantener una diuresis > 3 l/día), prevención de la hiperuricemia con alopurinol, dieta pobre en calcio y alcalinización de la orina con bicarbonato (oral o intravenoso) en caso de terapia intensiva con citostáticos. Se debe prevenir la insuficiencia renal evitando en lo posible los fármacos nefrotóxicos y tratando precozmente las infecciones y la hipercalcemia.
Prevención de las complicaciones óseas: se recomienda la movilización y deambulación evitando el encamamiento y el reposo prolongado para contrarrestar la pérdida de masa ósea, habida cuenta de que la patología ósea es una importante causa de morbilidad y mortalidad en pacientes con MM. Desde el inicio, en todos los pacientes en tratamiento, tanto en presencia como en ausencia de osteopenia o lesiones líticas, se administrarán inhibidores de resorción ósea como son los bisfosfonatos. Se pautarán unos 24 meses desde el diagnóstico y posteriormente en caso de recidiva o nueva actividad del proceso.
En este sentido, un amplio meta-análisis de la Cochrane (Mhaskar et al., 2017) concluyó que el uso de bisfosfonatos en pacientes con MM reduce notablemente la incidencia de fracturas vertebrales patológicas y el dolor óseo, pero se asocian con un riesgo incrementado de padecer osteonecrosis de la mandíbula (en 1 de cada 1.000 pacientes). Si bien no se concluyó sobre la superioridad clínica para ninguno de los fármacos disponibles, parece que el zoledronato es el fármaco que podría tener un mejor resultado en supervivencia global y en la incidencia de fracturas vertebrales. Es, pues, el agente más empleado y recomendado por las guías clínicas (Anderson et al., 2018): una pauta de al menos 2 años de duración con zoledronato por vía intravenosa a dosis única de 4 mg/mes; como alternativas eficaces, se pueden valorar el pamidronato (infusión mensual de 90 mg) o denosumab, el cual parece tener una menor toxicidad renal que zoledronato y podría ser preferible en pacientes con insuficiencia renal.
Analgesia: el dolor óseo gravitativo es el síntoma más frecuente en estos pacientes. El mejor tratamiento del dolor es el del propio MM, pero conviene añadir una pauta de analgesia enérgica, preferiblemente por vía oral y siguiendo la escalera terapéutica propuesta por la OMS, esto es, seleccionando analgésicos no opioides en primer lugar, pasando a opioides (opioides menores, y después, mayores) si se observa ineficacia. El empleo de esteroides y bisfosfonatos muestra a medio plazo una respuesta favorable analgésica.
Tratamiento de la anemia: la eritropoyetina (epoetina alfa o beta, o darboepoetina) es muy eficaz en el MM.
Tratamiento de complicaciones:
Hipercalcemia: se observa en un 30% de los pacientes. A priori, con hiperhidratación y los nuevos bisfofonatos por vía intravenosa, la corrección de esta complicación es rápida; se administrará 1 dosis de 4 mg de zoledronato, controlando los niveles de Ca2+ por si se precisa de una segunda o tercera dosis.
Hiperviscosidad: se observa en un 5-10% de los pacientes. Suele ocurrir con cantidades muy elevadas de componente monoclonal en suero, sobre todo si es MM IgM que no sea macroglobulinemia o con polímeros de IgA. Se maneja con hidratación e inicio de la quimioterapia para reducir la producción del componente M, además de plasmaféresis y recambio plasmático. En pacientes con factores con riesgo de trombosis venosas periférica se suele recurrir a tromboprofilaxis temporal (durante al menos 4-6 meses, dependiendo del riesgo), bien con ácido acetilsalicílico o con heparinas de bajo peso molecular (enoxaparina).
Complicaciones neurológicas: ocurre lesión medular espinal o radicular en el 5-10% de los casos, por lo general debido a la invasión del canal medular por plasmocitomas de localización en vértebras adyacentes o expansión paravertebral; las medidas recomendadas son: radioterapia, corticoides y laminectomía si fuera necesario. Además, el control de la neuropatía periférica en pacientes con MM requiere evitar o extremar precauciones con el uso de fármacos potencialmente neurotóxicos (bortezomib, talidomida); el dolor neuropático superficial puede tratarse con lidocaína tópica al 5%, recurriendo en casos no controlados a tratamientos con amitriptilina, pregabalina, duloxetina o tramadol.
El futuro del tratamiento
A pesar de los avances terapéuticos, el MM se sigue considerando una enfermedad incurable, fundamentalmente debido al desarrollo de resistencias: una vez que los pacientes se vuelven resistentes a un determinado tratamiento, la esperanza de vida se reduce notablemente. Por ello, resulta necesaria la investigación farmacológica que permita diseñar fármacos y terapias avanzadas con nuevos mecanismos de acción para ser empleados, en una primera instancia, en pacientes refractarios a fármacos “convencionales”. Después, si su balance beneficio-riesgo se muestra muy favorable, podrán avanzar en las líneas de tratamiento hasta convertirse en opciones de primera línea, como fue el caso de daratumumab. Afortunadamente, la investigación biomédica en MM, especialmente en pacientes difíciles de tratar (en el contexto de recaída y/o refractariedad), es bastante profusa13. En los últimos años y meses se han divulgado resultados clínicos para novedosos fármacos y estrategias terapéuticas; algunos de los más prometedores se comentan a continuación.
Recientemente, la EMA ha autorizado en Europa isatuximab (Sarclisa®), el cual aún no está disponible en España. Se trata de un anticuerpo monoclonal de administración por infusión intravenosa que se une a un epítopo extracelular específico del receptor CD38 causando la muerte de la célula mielomatosa por un mecanismo de acción como el de daratumumab. Se ha aprobado, en combinación con pomalidomida y dexametasona, en la indicación de MM recidivante y refractario en pacientes adultos cuya enfermedad ha progresado tras al menos 2 líneas de tratamiento previas incluyendo lenalidomida y un inhibidor de proteasoma.
—La recaída es la mayor complicación del tratamiento y requiere el uso precoz y secuencial de regímenes de rescate—
La aprobación se ha hecho en base a los resultados del ensayo clínico pivotal de fase 3 ICARIA-MM (Attal et al., 2019), en el que el triplete con isatuximab redujo significativamente el riesgo de progresión o muerte en un 40% en comparación con la asociación de pomalidomida con dexametasona, prolongando la mediana de SLP hasta casi 1 año (11,5 meses), lo que supone una mejora de 5 meses respecto al estándar de tratamiento; las tasas de respuesta global también fueron significativamente superiores con el triplete (60% vs. 35%). El beneficio clínico de la adición de isatuximab se demostró en pacientes > 75 años, con insuficiencia renal y con refractariedad a lenalidomida. En términos de seguridad, las reacciones adversas más frecuentemente descritas con isatuximab fueron: neutropenia (47%), reacciones relacionadas con la infusión, neumonía e infecciones de las vías respiratorias altas (28-31%) o diarrea (26%); por su gravedad, sobresalen la neumonía y la neutropenia febril.
Más reciente han sido los resultados provisionales del ensayo pivotal IKEMA que se han presentado durante la sesión de ‘Últimas novedades’ del Congreso Virtual de la Asociación Europea de Hematología, del 14 de junio de 2020. Según dichos datos, la adición de isatuximab a carfilzomib y dexametasona redujo en un 47% el riesgo de progresión de la enfermedad o muerte respecto al estándar de tratamiento con el doblete de fármacos, en pacientes con MM recidivante (con 1-3 terapias previas). Aunque sin diferencias en la tasa de respuesta global (87% vs. 83%), la respuesta a la triple terapia fue considerablemente más profunda, con niveles indetectables de enfermedad (respuesta completa) en casi el 30% (29,6% vs. 13%). En el momento del análisis provisional, los datos de SG eran aún inmaduros. Los datos de tolerabilidad del nuevo fármaco estaban en línea con el perfil de seguridad descrito en otros estudios, sin nuevos hallazgos destacables.
Otra opción en fase de investigación es belantamab mafodotin, un fármaco basado en la conjugación de un anticuerpo monoclonal con una molécula citotóxica, que sigue la vía terapéutica iniciada por otros agentes antineoplásicos aprobados en la última década (como brentuximab vedotina o trastuzumab emtansina), dirigidos frente a biomarcadores tumorales específicos para ejercer la citotoxicidad sobre una población concreta de células. Así, se trata de un inmunoconjugado formado por una IgG humanizada anti-BCMA unida a través de un conector resistente a proteasas (maleimidocaproil) a un agente citotóxico anti-microtúbulos, la monometil auristatina F. La proteína BCMA o antígeno de maduración de las células B (B-cell maturation antigen), también denominado CD269 o receptor de TNF de la superfamilia 17 (TNFRSF17), es un receptor expresado en la superficie celular casi exclusivamente en células del linaje B, sobre todo en el estadio de diferenciación de células B maduras a células plasmáticas (por tanto, en muchas células de MM), pero está virtualmente ausente en células B naïve y de memoria o en células madre hematopoyéticas, lo cual le convierte en una diana terapéutica selectiva.
Belantamab mafodotin fue evaluado en un estudio multicéntrico y multinacional de fase 2 (DREAMM-2), con diseño abierto y de 2 brazos (2 dosis), en el que se aleatorizaron un total de 196 pacientes adultos con MM recidivante o refractario y relativo buen estado funcional (estado ECOG 0-2), quienes habían recibido ≥ 3 líneas de tratamiento (incluyendo inmunomoduladores, inhibidores de proteasoma y un anti-CD38). Los resultados publicados tras el análisis intermedio por intención de tratar (Lonial et al., 2019) demuestran que la tasa de respuesta objetiva general según revisión central independiente fue del 31% y el 34% en los brazos 1 y 2 (tratados con 2,5 y 3,4 mg/kg, 1 ciclo i.v. cada 3 semanas), respectivamente. Son respuestas clínicamente relevantes en situaciones con escasas opciones de tratamiento, que determinan un balance beneficio-riesgo potencialmente positivo, a pesar de que el perfil toxicológico es importante, caracterizado por la aparición frecuente de eventos adversos de grado 3-4 (40-47% de pacientes), tales como queratopatía (21-27%), trombocitopenia (20-33%) y anemia (20-25%), e incluso dos muertes posiblemente relacionadas con el tratamiento.
Los resultados preliminares que han sido publicados en el Congreso Anual de la Sociedad Americana de Oncología Clínica14 (ASCO), celebrado en mayo de 2020, refuerzan el potencial del belantamab mafodotin en pacientes con MM en recaída o refractario. Una actualización a los 13 meses del estudio DREAMM-2 indica que la dosis más baja del fármaco aporta una mediana de supervivencia global de 15 meses y de 11 meses para la duración de la respuesta, en una población de pacientes con una media de 7 líneas de tratamiento, refractarios a muchos fármacos; más de 1 de cada 3 pacientes (36%) logran un beneficio clínico (respuesta mínima o superior). Por otra parte, los resultados iniciales del estudio de fase 1/2 DREAMM-6 sugieren que la combinación de belantamab mafodotin con bortezomib y dexametasona aporta una tasa de respuesta objetiva del 78% (50% de respuesta muy buena) en pacientes en recaída tras 1 o más líneas de tratamiento. La seguridad y tolerabilidad del nuevo fármaco concuerdan con los resultados previamente publicados.
Por tanto, aunque aún se requieren datos derivados de ensayos controlados de fase 3, a día de hoy la solicitud de autorización de comercialización de belantamab mafodotina ya está bajo revisión por parte de la FDA estadounidense y es previsible que la EMA europea pueda seguir estos pasos en un futuro próximo si se confirman estos positivos resultados.
Finalmente, hay que hacer una mención especial al recorrido que los medicamentos CAR-T pueden tener en el tratamiento y la curación del MM, tras la autorización en Europa en 2019 de los primeros fármacos anti-CD-19 de fabricación industrial de este tipo de terapia avanzada: tisagenlecleucel y axicabtagén ciloleucel (Revuelta et al., 2019). En la actualidad, se están desarrollando al menos 15 ensayos clínicos a nivel internacional con candidatos de células T –extraídas del propio paciente– modificadas para expresar un receptor de antígeno quimérico o CAR, que están revelando algunos resultados prometedores en MM (para una información más profunda sobre este tema, se recomienda consultar la revisión de Lin et al., 2019).
Entre los diversos candidatos estudiados, la mayoría consisten en células T que expresan CAR específicamente dirigidos para reconocer el antígeno BCMA, algunos de los cuales son de última generación y expresan diversos motivos coestimuladores; incluso uno de ellos aporta el aspecto innovador de no presentar la estructura de inmunoglobulina y producirse a través de un mecanismo de transfección que no utiliza vectores virales, sino basado en trasposones. Las células CAR-T anti-BCMA han demostrado, en ensayos de fase 1 (solo dos candidatos han llegado a fase 1/2) con pacientes con MM en recaída-refractariedad ampliamente pre-tratados y de alto riesgo, elevadas tasas de respuestas objetivas, en el rango de 81-100%, y con respuestas completas en hasta el 68% de pacientes en algún estudio; respuestas que aparecían a los 14-30 días post-infusión y para las que en algún paciente se ha descrito una duración –dosis-dependiente en muchos casos– de más de 10 meses. En línea con lo ya conocido sobre la toxicidad de estas terapias, destaca la frecuencia de aparición del síndrome de liberación de citocinas y eventos de neurotoxicidad (encefalopatía), que en principio pueden ser manejados clínicamente. Podría ser previsible que algún medicamento a base de células CAR-T anti-BCMA pueda ser aprobado en el corto plazo y, a expensas de mejoras en otras líneas de tratamiento, quizá ser la mejor opción para alcanzar la curación del MM a medio plazo.
Otros candidatos de células CAR-T dirigidos a la proteína CD138, a la proteína SLAMF7 o frente a cadenas ligeras también parecen mostrar resultados clínicos prometedores. Incluso la administración de tisagenlecleucel (anti-CD19) tras un trasplante autólogo de progenitores hematopoyéticos, o la estrategia de células CAR-T dual específicas para reconocer BCMA y CD19 a la vez están mostrando actividad interesante en MM en recaída-refractariedad. Pero se trata aún de fases precoces de la investigación clínica: los prometedores resultados requieren ser confirmados en ensayos de eficacia y seguridad –de al menos fase 2– con una muestra significativa de pacientes.
El papel asistencial del farmacéutico
Todos los profesionales farmacéuticos, desde sus diversos ámbitos profesionales y de competencias, pueden contribuir al adecuado asesoramiento y asistencia sanitaria a los ciudadanos y pacientes con mieloma múltiple. Teniendo en cuenta las particularidades de la enfermedad comentadas en la presente revisión y el hecho de que la práctica totalidad de medicamentos empleados en su tratamiento son calificados como de uso –y dispensación– hospitalario, la figura del farmacéutico especialista a nivel de hospital cobra una especial relevancia en la consecución de los objetivos del proceso farmacoterapéutico, desde la contribución a la selección del régimen y pauta de tratamiento óptimos hasta la monitorización del paciente para evaluar ajustes posológicos y de tratamientos concomitantes (Fajardo et al., 2016).
No obstante, a pesar de que puedan requerir ingresos hospitalarios frecuentes, todos los pacientes con diagnóstico de MM van a estar en tratamiento crónico la mayor parte del tiempo en el ámbito ambulatorio y, con seguridad, van a ser tratados con más de un fármaco (polimedicados), tanto para el tratamiento del propio mieloma como para el manejo de otros síntomas y signos; así, la adherencia terapéutica representa un pilar fundamental del tratamiento, con influencia en la calidad de vida de los pacientes. En ese contexto, la proximidad y accesibilidad del farmacéutico comunitario para los pacientes y ciudadanos permite que también pueda ejercer una labor asistencial proactiva a través de los Servicios Profesionales Farmacéuticos Asistenciales.
Atendiendo al hecho de que cada día más dos millones de pacientes y usuarios acuden a las más de 22.000 farmacias españolas, y que en ellas se ofrecen al año más de 182 millones de consejos sanitarios, parece evidente el enorme potencial divulgador del farmacéutico como profesional sanitario, así como su incuestionable papel para canalizar hacia el médico a personas con problemas relevantes de salud, para un estudio clínico detallado. En definitiva, la oficina de farmacia constituye un establecimiento sanitario accesible (con amplitud de horarios y sin necesidad de cita previa) y ubicuo capaz de suministrar una información rigurosa, de ofrecer un servicio de máximas garantías sanitarias con la debida confidencialidad, y de promover un mejor uso de los medicamentos a fin de prevenir los problemas relacionados con los mismos; facilita, además, la disponibilidad de los medicamentos usados en el tratamiento de soporte del MM, con claras implicaciones en la sostenibilidad del Sistema Nacional de Salud.
Con la integración efectiva del farmacéutico en los equipos multidisciplinares de atención primaria y hospitalaria, se pueden identificar varias vías asistenciales de actuación para con los pacientes con MM.
Educación sanitaria
El farmacéutico puede y debe aportar a los pacientes información con rigor científico sobre:
los medicamentos: incidiendo sobre su objetivo y mecanismo, las peculiaridades de conservación (si las hubiera), el momento óptimo de administración, la posibilidad e importancia de interacciones con otros medicamentos (incluidos los de automedicación), etc.
su pauta de administración: se puede aconsejar la adaptación de la toma coincidiendo con eventos cotidianos o aportar diagramas que ayuden a relacionar la medicación con hábitos de vida.
A la hora de afrontar una enfermedad como el MM, es importante transmitir, con la sensibilidad adecuada, que se trata de un proceso crónico e incurable, en la que solo un porcentaje bajo de pacientes se considerarán virtualmente curados. No obstante, aunque los tratamientos contra el MM tienen una toxicidad no desdeñable, no impide que los pacientes lleven una vida prácticamente normal, con una calidad de vida bastante aceptable durante varios años. En este sentido, la información rigurosa y el asesoramiento práctico son piezas clave para combatir un tipo de cáncer en expansión epidemiológica.
Se debe advertir al paciente de que, aun con un tratamiento exitoso, convivirán con un riesgo incrementado de fracturas óseas –de lenta recuperación– por una fragilidad aumentada; resulta especialmente importante la prevención de aplastamientos vertebrales, evitando levantar o soportar pesos importantes. En general, se recomienda que los pacientes con MM mantengan la mayor actividad física posible, para lo cual es fundamental lograr un adecuado control del dolor. El ejercicio moderado refuerza la musculatura y favorece la recalcificación ósea. Por el riesgo incrementado de fracturas, no se recomiendan deportes de contacto o aquellos que impliquen movimientos bruscos y violentos. Con respecto a los cuidados nutricionales, se recomiendan los alimentos ricos en calcio (como queso y otros lácteos), pues, a pesar de que los pacientes suelan presentar inicialmente hipercalcemia, el común tratamiento con bisfosfonatos –favorecen la recalcificación mediante la captación de calcio de sangre– hace que la calcemia disminuya; la dieta rica en calcio es la medida principal, si bien puede valorarse el uso de suplementos de calcio y vitamina D.
Por otra parte, conviene recordar a pacientes y familiares que el MM implica un riesgo incrementado de padecer infecciones: se estima casi 10 veces superior al de un sujeto sano, más alto incluso cuando la enfermedad no está controlada, en fases avanzadas o en la etapa inicial de cualquier tratamiento. Las infecciones de las vías respiratorias son las más frecuentes y, potencialmente, las más graves, por lo que se pueden recomendar ciertas medidas básicas para reducir el número de episodios: evitar el contacto directo con familiares o personas sintomáticas, extremar la higiene o evitar en la medida de lo posible las aglomeraciones en épocas de gripe. Tal y como se está repitiendo en la pandemia por COVID-19, el lavado frecuente de manos también adquiere relevancia en determinadas situaciones (por ejemplo, salas de espera de hospitales), y en momentos de especial susceptibilidad (por ejemplo, tras un trasplante) puede ser recomendable portar una mascarilla quirúrgica o filtrante que evite la transmisión por el aire de microorganismos patógenos. Otras medidas sencillas para minimizar la probabilidad de infecciones son: una nutrición sana, mediante una dieta variada y equilibrada15, una buena hidratación, ejercicio moderado y evitar el tabaco o las aglomeraciones en ambientes cerrados.
Además, desde la farmacia se puede ejercer una acción social crucial, mediante la orientación de los pacientes con nuevo diagnóstico de MM para que se enrolen en asociaciones de pacientes que les brindarán acompañamiento, apoyo y diversos servicios socio-sanitarios. Dos buenas opciones son, por ejemplo, el foro de pacientes y ex-pacientes de la Fundación Josep Carreras o la Comunidad Española de Pacientes de Mieloma Múltiple (CEMM).
—Los farmacéuticos, en sus distintos ámbitos de actuación, juegan un papel clave en la optimización de los resultados clínicos de la farmacoterapia—
Detección precoz
Habida cuenta de que muy pocos casos de MM están relacionados con factores de riesgo evitables (a diferencia de otros tipos de cáncer que sí tienen mayor relación con determinados factores), se considera que en la actualidad no se puede hacer nada para evitar la aparición del mieloma. Las personas con una Gammapatía Monoclonal de Significado Incierto son más propensas a desarrollarlo y se recomienda su monitorización más estrecha, si bien todavía no está claro cuál es el mecanismo que hace que unas personas desarrollen MM y otras no. Por ello, cobra mayor relevancia un diagnóstico rápido que permita un tratamiento temprano, pues el momento de inicio es un factor decisivo en la eficacia del mismo.
La detección precoz del mieloma es difícil porque los síntomas no son específicos y se parecen a los de otras afecciones, y porque a menudo no debutan las manifestaciones hasta fases avanzadas. Sin embargo, puede ser detectado en un análisis rutinario debido a un nivel inusualmente alto de proteínas en la sangre (proteínas totales). También se debe sospechar de posible MM ante un bulto o tumor de consistencia dura que normalmente crece a partir de una zona de hueso como el cráneo, una costilla o el esternón. Si, por el contrario, las masas de mieloma o plasmocitomas se generan en las vértebras, son más difícilmente visibles o palpables (suelen crecer hacia el interior), pero pueden provocar la compresión de la médula espinal o las raíces de los nervios que salen de ella, con manifestaciones graves como la pérdida de fuerza o de sensibilidad de las piernas o la pérdida de control de los esfínteres. Aunque estos últimos signos son poco frecuentes, su detección a nivel de la farmacia comunitaria requiere de la derivación urgente al médico para un diagnóstico preciso.
Hay que tener en cuenta que en personas de edad avanzada el motivo más frecuente de dolor óseo son los trastornos degenerativos (como la artrosis) y las causas más frecuentes de fatiga se achacan a patologías cardiorrespiratorias, todos ellos mucho más frecuentes que el MM. No obstante, la existencia de dolor que no se controla con los tratamientos analgésicos habituales y casos de fatiga persistente que aumenta con el tiempo pueden también considerarse señales de alerta sugerentes de MM. Un aplastamiento vertebral, por ejemplo, aunque se produzca en una persona mayor, debe conducir a un estudio básico analítico y radiológico básico, que permitiría detectar la presencia de anemia, proteínas por encima de los niveles normales, alteración de la función renal, aumentos del calcio en sangre o bien lesiones líticas o pequeñas fracturas óseas no achacables a un traumatismo previo; hallazgos que guían al posible diagnóstico de MM y hacen necesaria la derivación del paciente al médico especialista.
Optimización de la terapia farmacológica
Una vez establecido el diagnóstico de MM y el tratamiento por el hematólogo/oncólogo, el farmacéutico, como profesional sanitario experto en el medicamento, debe velar por el uso seguro y eficaz de los mismos, para que los pacientes alcancen el máximo beneficio clínico. Esto es aplicable tanto en el entorno hospitalario, donde acudirán los pacientes a recibir los ciclos de quimioterapia o a la realización de un trasplante de progenitores hematopoyéticos, como, con igual o mayor relevancia, en el ámbito comunitario, pues hay que recordar que el farmacéutico comunitario conoce toda la medicación que utiliza un paciente ambulatorio, no solo la medicación prescrita frente al mieloma, sino también los tratamientos para enfermedades concomitantes, medicamentos que no necesitan prescripción, el uso de complementos alimenticios, etc.
En el momento de la dispensación de cualquier medicamento prescrito en el curso de la enfermedad, el farmacéutico comprobará que el paciente cuente con toda la información necesaria para que el uso del mismo sea efectivo y seguro. Para ello, es conveniente averiguar si existe algún criterio que impida la dispensación, por ejemplo, alergia a algún componente del medicamento, una contraindicación absoluta o interacciones con otros medicamentos (o alimentos), una duplicidad o una situación fisiológica especial como puede ser el embarazo o lactancia. Si es la primera vez que esa persona va a utilizar dicho medicamento, la labor del farmacéutico será asegurar que la persona sale de la farmacia conociendo para qué es ese medicamento y cuál es su correcto proceso de uso. Si no fuera la primera vez (dispensación de continuación), el farmacéutico evaluará si dicho medicamento está siendo eficaz y seguro, fundamentalmente verificando si ha habido cambios en el tratamiento (dosis, pauta posológica, duración del tratamiento, adición de nuevos medicamentos, etc.) y si el paciente ha experimentado algún problema con el tratamiento que pudiera hacer sospechar de una posible reacción adversa, interacción, contraindicación, etc. Además de los medicamentos prescritos, en muchas ocasiones el propio paciente solicitará consejo al farmacéutico sobre los diferentes síntomas que van apareciendo.
Como en tantas enfermedades que requieren tratamiento crónico, al menos tras los diversos episodios de recaída, la adherencia terapéutica ha sido descrita como uno de los factores de mayor influencia sobre los resultados de la farmacoterapia del MM. Si bien los pacientes suelen cumplir adecuadamente los ciclos de medicación anti-mieloma (son conscientes de la gravedad de la enfermedad y de la importancia del cumplimiento del tratamiento, pues acuden al hospital para ello), con niveles de adherencia superiores al 70% –y hasta cercanos al 100%– en la mayoría de fármacos más frecuentemente empleados (Cransac et al., 2019), la promoción de la adherencia a los medicamentos usados para el tratamiento de soporte es una de las facetas que pueden reforzarse desde la farmacia comunitaria. Las estrategias para asegurar que el paciente se involucre voluntariamente en su tratamiento deben desarrollarse de forma personalizada, con el paciente y la familia, fomentando su confianza en los fármacos administrados; pueden incluir información verbal y escrita y recursos interactivos, debiendo siempre recordarse que las consecuencias de la falta de adherencia pueden ir desde un empeoramiento de la calidad de vida, una falta de control de la enfermedad y una mayor probabilidad de complicaciones, hasta la aparición de efectos secundarios o incluso de mortalidad. Todo ello, además, puede suponer ingresos hospitalarios adicionales y otras intervenciones sanitarias que impliquen ineficiencia del gasto farmacéutico y sanitario.
A este respecto, un reciente estudio demostró que la implicación activa del farmacéutico resulta beneficiosa en la terapia de pacientes con mieloma. Con servicios como la revisión de la medicación y su adecuación a las guías clínicas o el manejo de los efectos adversos relacionados con medicamentos, se demostró –en comparación con la ausencia de intervención de un farmacéutico– una mejora significativa en la adherencia a los tratamientos de soporte, como bifosfonatos (96% vs. 68%), calcio y vitamina D (100% vs. 41%), aciclovir (100% vs. 58%) y profilaxis frente a la neumonía por Pneumocystis jirovecii (100% vs. 50%).
Por otra parte, tras una dispensación de inicio o de continuación, y especialmente en los tratamientos prolongados, un adecuado seguimiento farmacoterapéutico (ofrecido por el farmacéutico de forma rutinaria, sistematizada y registrada/documentada, con reuniones periódicas con el paciente) permitirá detectar, atenuar y resolver la posible aparición de resultados negativos y problemas relacionados con la farmacoterapia. La farmacovigilancia ante posibles reacciones adversas (con su correspondiente notificación, en su caso, al Sistema Nacional de Farmacovigilancia), y la identificación y prevención de interacciones farmacológicas y contraindicaciones del tratamiento antineoplásico y de soporte revertirán en una mejor calidad de vida de los pacientes con MM. Además, mediante una actitud vigilante, la detección precoz desde la oficina de farmacia o la farmacia hospitalaria de los signos y síntomas que acompañan a una posible recaída o refractariedad a una línea de tratamiento y a las potenciales complicaciones derivadas de la inmunosupresión (infecciones, tumores, osteoporosis, etc.) también puede contribuir a activar la ruta asistencial que asegure un cambio de tratamiento temprano.
Para todo ello, además de lo especificado en el presente informe y de la recomendación de consultar las fichas técnicas autorizadas de los medicamentos, si se tiene en consideración que la información científica se actualiza constantemente y que en MM se han aprobado numerosas alternativas terapéuticas en los últimos años, cobran especial relevancia las bases de datos que contienen información actualizada y pormenorizada sobre aspectos farmacológicos. Es el caso, por ejemplo, de la base de datos BOT PLUS, que permite, entre otras funcionalidades, la detección de interacciones farmacológicas entre múltiples medicamentos comerciales y/o principios activos, para su evaluación a la hora de optimizar los tratamientos.
Por último, conviene recordar y tener presente algunos conceptos sobre seguridad de los tratamientos usados frente al MM, entre los que podemos destacar:
En pacientes sometidos a autotrasplante de progenitores hematopoyéticos, desde el inicio del acondicionamiento y durante casi 15-21 días posteriores, pueden producirse diversos efectos secundarios (náuseas, vómitos, mucositis oral, fiebre, infecciones o hemorragias), cuya intensidad estará relacionada con la intensidad del tratamiento de acondicionamiento; durante ese periodo, además de terapia preventiva y de soporte, puede ser necesario administrar nutrición parenteral.
Las citopenias ocurren con no poca frecuencia como eventos adversos relacionados con las terapias actuales contra el mieloma, incluidos los fármacos alquilantes y los nuevos agentes inhibidores del proteasoma o inmunomoduladores. Con estos últimos, se ha reportado anemia de grado ≥ 3 en proporciones significativas de pacientes (desde el 3% al 19%) y, por tanto, se deben considerar los agentes estimulantes de la eritropoyesis y la suplementación óptima con hierro si la anemia no mejora con la quimioterapia.
La trombocitopenia es común con inhibidores del proteasoma como bortezomib y carfilzomib, así como con fármacos inmunomoduladores (talidomida, lenalidomida y pomalidomida), por lo que la reducción de la dosis debe realizarse en consecuencia y el tratamiento debe interrumpirse en caso de trombocitopenia de grado 4. La neutropenia es también un evento adverso frecuente con los fármacos inmunomoduladores y el anticuerpo monoclonal daratumumab, con una incidencia cada vez mayor en el contexto de recaída y en la terapia combinada. Por tanto, en pacientes considerados de alto riesgo de neutropenia febril, se recomienda el uso del factor estimulante de colonias de granulocitos.
En pacientes con insuficiencia renal, es crucial seleccionar la terapia adecuada: bortezomib y talidomida pueden administrarse sin ningún ajuste de dosis, mientras que el ajuste de la dosis debe ser cuidadoso cuando se inicia lenalidomida o pomalidomida. Bortezomib tiene la ventaja adicional de eliminación rápida de las cadenas ligeras libres, acelerando así la respuesta renal.
Para una mayor información sobre los riesgos de seguridad de las nuevas terapias contra el mieloma, así como posibles opciones de manejo de las complicaciones de las mismas, se recomienda consultar la detallada revisión de McCullough y colaboradores (McCullough et al., 2018). De cara al paciente, es aconsejable e interesante la consulta del documento “Recomendaciones básicas para pacientes en relación a la toxicidad por quimioterapia” de GEPAC (Grupo Español de Pacientes con Cáncer), disponible en:
Esta es la pregunta que se han venido haciendo muchos expertos y gran parte de la sociedad en los últimos meses. Y, aunque hay grandes incertidumbres al respecto, un reciente estudio (Bhardwaj et al., 2020) ha arrojado cierta luz sobre el asunto. Los autores han pretendido identificar cuáles son los factores que determinan la persistencia de los virus en el aire y en superficies: para ello, han evaluado comparativamente el tiempo de secado de las gotículas respiratorias (en las que se sabe se “transporta” el nuevo coronavirus y actúan como vehículo de contagio entre personas) en seis ciudades de todo el mundo (Nueva York, Chicago, Los Ángeles, Miami, Sidney y Singapur), con diferentes condiciones climatológicas y diferente evolución de la pandemia de COVID-19. Emplearon un modelo de evaporación de difusión limitada para una gotícula colocada en una superficie parcialmente mojada (entre ellas, las más recurrentes, como pomos de puertas o pantallas de móviles) y con una línea de contacto fijada. Mediante una serie de modelos matemáticos, calcularon las posibilidades de supervivencia del virus en la gotícula en función de la vida útil de las mismas bajo distintas condiciones ambientales, considerando también la variación en el volumen de la gota y el ángulo de contacto.
Sus resultados sugieren que el tipo de superficie, la humedad relativa y la temperatura ambiente determinan notablemente las posibilidades de supervivencia, siendo estas mayores cuanto mayor es la humedad (hasta 5 veces más respecto a ambientes secos) y menor la temperatura ambiente, pues temperaturas más altas contribuyeron a secar la gotícula con mayor rapidez y redujeron drásticamente las posibilidades de persistencia del virus. Esto explica que el tiempo de secado de una gotícula de 5 nl en Nueva York (temperatura ambiente en marzo-abril de 6-10ºC y humedad relativa 50-60%) era de casi 90 s, mientras que, en el otro extremo, ese tiempo se reducía a menos de 30 s en Singapur (28-32ºC y 70-80% de humedad), lo cual puede correlacionarse con la mayor y menor tasa de crecimiento de las infecciones por SARS-CoV-2 de entre las ciudades consideradas, aunque no exclusivamente (influyen otros factores, principalmente las medidas preventivas y condiciones ambientales en espacios cerrados). Además, el estudio sugiere que las superficies como las pantallas de los teléfonos móviles, el algodón y la madera deben limpiarse con mayor frecuencia que las superficies de vidrio y acero, ya que estas últimas son relativamente hidrófilas y, en ellas, las gotículas se evaporan más rápido.
En definitiva, parece que, en lugares con climas más cálidos y secos, como puede ser gran parte del interior de España en verano, la permanencia de las gotículas respiratorias en el ambiente y superficies se puede acortar, y podría favorecer la mitigación de los contagios. No obstante, no hay suficiente información para descartar una segunda ola epidémica, y se deben extremar las medidas profilácticas, también en verano. A este respecto, los autores de otro trabajo reciente (Zhang et al., 2020) advierten de que, si bien el distanciamiento social y el lavado de manos deben continuar, son probablemente insuficientes, y el uso de mascarillas se revela como la medida más efectiva para evitar contagios interpersonales, aunque al inicio de la pandemia hubiera cierta controversia en las recomendaciones de su uso. También mediante diversos modelos matemáticos, concluyeron que el uso generalizado de mascarillas redujo, por sí mismo, el número de infecciones en 78.000 casos en Italia (desde el 6 de abril al 9 de mayo) y en más de 66.000 en Nueva York (desde el 17 de abril al 9 de mayo). En esa misma línea, un tercer estudio (Stutt et al., 2020), consistente en un modelo matemático que contempla una simulación con 60 millones de personas, sugería que el uso continuado de mascarillas faciales en público por al menos la mitad de la población puede reducir al mínimo la tasa de reproducción del virus (Ro < 1), siempre que no se relajen el resto de medidas preventivas.
Bhardwaj R, Agrawal A. Likelihood of survival of coronavirus in a respiratory droplet deposited on a solid surface. Phys Fluids (1994). 2020; 32(6): 061704. DOI: 10.1063/5.0012009.
Stutt RO, Retkute R, Bradley M, Gilligan CA, Colvin J. A modelling framework to assess the likely effectiveness of facemasks in combination with ‘lock-down’in managing the COVID-19 pandemic. Proceedings of the Royal Society A. 2020; 476(2238): 20200376.
Zhang R, Li Y, Zhang AL, Wang Y, Molina MJ. Identifying airborne transmission as the dominant route for the spread of COVID-19. Proc Natl Acad Sci U S A. 2020; 202009637. DOI: 10.1073/pnas.2009637117.
En 2019, la Organización Mundial de la Salud (OMS) alertó de que la prevalencia de resistencias a los fármacos de primera línea frente al VIH-1 (especialmente los regímenes a base de inhibidores de la transcriptasa inversa no nucleosídicos) está en crecimiento y, en algunos países con escasas opciones terapéuticas, más del 10% de las nuevas infecciones por VIH se producen ya con virus que han desarrollado resistencias a varios fármacos.
En este sentido, un reciente artículo describe el caso de un varón de raza caucásica de 80 años de edad que fue diagnosticado de infección por VIH-1 en 1989 (a los 41 años de edad) e inició el tratamiento antirretroviral en 1995; desde entonces, ha recibido más de 14 fármacos diferentes, habiendo experimentado el último fracaso virológico a un régimen basado en dolutegravir. En noviembre de 2017, los autores del trabajo recolectaron una muestra de sangre del paciente e identificaron una cepa del VIH-1 del subtipo B. Se analizaron las resistencias tanto a nivel genotípico (usando el algoritmo de interpretación de la Base de Datos de Resistencia a Fármacos antirretrovirales de Stanford) como fenotípico (usando los ensayos PhenoSense y Trofile). El virus demostró una amplia resistencia cruzada genotípica y fenotípica a todos los fármacos anti-VIH aprobados en Europa, incluidos los inhibidores de integrasa de segunda generación más modernos, como dolutegravir y bictegravir. Si bien es cierto que la literatura científica ya había registrado casos con resistencias a algunos de los fármacos de cada grupo (pero no a todos simultáneamente), se trata del primer caso descrito de una persona infectada cuyo virus es “panresistente”, esto es, resistente a las 5 familias de fármacos orales y a todas sus combinaciones que se usan habitualmente contra el VIH: inhibidores de la transcriptasa reversa análogos de nucleósidos y no nucleosídicos, inhibidores de la proteasa, inhibidores de la integrasa e inhibidores de la fusión. El virus se mostró insensible a 25 de los 26 fármacos probados, y exclusivamente fue sensible a tenofovir, que por sí solo no va a ser eficaz contra el virus.
Así pues, sin opciones terapéuticas a excepción de los fármacos en investigación, este caso es un claro ejemplo de cómo, a pesar del amplio número de opciones terapéuticas disponibles, aún se necesitan nuevos antirretrovirales con diferentes mecanismos de acción. Las resistencias cruzadas del VIH siguen siendo una preocupación y una amenaza importantes (de especial relevancia en pacientes que han sido tratados con combinaciones subóptimas de antirretrovirales en el pasado) para erradicar la pandemia del SIDA, pues comprometen los resultados clínicos, dificultan el manejo terapéutico y aumentan el riesgo de transmisión. Este trabajo es otra muestra del gran impacto en salud pública que representan las resistencias antimicrobianas y de que debe aumentar la conciencia sobre la necesidad de optimizar el uso y la adherencia a fármacos antimicrobianos (tanto antivirales como antibióticos).
Puertas MC, Ploumidis G, Ploumidis M, Fumero E, Clotet B, Walworth CM et al. Pan-resistant HIV-1 emergence in the era of integrase strand-transfer inhibitors: a case report. Lancet. 2020; DOI: https://doi.org/10.1016/S2666-5247(20)30006-9.
Ante la gran variabilidad en el comportamiento clínico de la infección por el nuevo coronavirus SARS-CoV-2, un grupo internacional de investigadores se propuso realizar un estudio genético a fin de determinar la potencial influencia de los genes en el desarrollo de la enfermedad. Para ello, incluyeron en el estudio 1.980 pacientes con confirmación diagnóstica de COVID-19 grave –definido por insuficiencia respiratoria y necesidad de oxígeno o ventilación mecánica– ingresados en 7 hospitales de Italia y España. Tras la exclusión de los pacientes con valores atípicos, restringieron la investigación a un total de 1.785 pacientes (y 2.205 participantes sanos como controles), de los cuales se analizaron más de 8 millones y medio de polimorfismos de nucleótido único a partir de muestras de sangre.
Un meta-análisis de los dos paneles de casos y controles identificó una mayor frecuencia de 26 variantes genéticas en los pacientes con fallo respiratorio, siendo de especial interés una potente asociación cruzada con el polimorfismo rs11385942 en el locus 3p21.31 (cromosoma 3) y con el polimorfismo rs657152 en el locus 9q34.2 (cromosoma 9), estadísticamente significativa a nivel del genoma; ambos polimorfimos se relacionaban con una probabilidad aumentada en un 77% (odds ratio u OR= 1,77; IC95% 1,48-2,11) y en un 32% (OR= 1,32; IC95% 1,20-1,47) de padecer insuficiencia respiratoria. Además, su frecuencia de aparición fue significativamente mayor en pacientes que requirieron ventilación mecánica respecto a aquellos que solo se les administró oxígeno, con independencia de edad y sexo de los pacientes.
De manera interesante, la asociación en el cromosoma 9 coincidió con el locus del grupo sanguíneo ABO: un análisis específico demostró un riesgo de gravedad de la COVID-19 un 45% mayor en pacientes con grupo sanguíneo A respecto a otros grupos sanguíneos (OR= 1,45; IC95% 1,20-1,75) y un efecto protector relacionado con el grupo sanguíneo O, con un 35% menos de riesgo de necesitar asistencia ventilatoria (OR= 0,65; IC95% 0,53-0,79); para el resto de grupos sanguíneos no parece haber una tendencia clara. No obstante, la relación más clara se encontró con la variante genética identificada en el cromosoma 3, que abarca 6 genes (SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6 y XCR1) y determina una mayor vulnerabilidad de los pacientes: uno de los genes se asocia con la estabilización de la enzima ECA2 (puerta de entrada del virus a las células humanas) y otros dos codifican para receptores de quimiocinas (implicadas en el proceso inflamatorio agudo –tormenta de citoquinas– a nivel pulmonar). Dicho polimorfismo fue más frecuente en personas de “menor” edad (media de 59 años) respecto a las más ancianas, lo que podría explicar al menos en parte los casos de mayor gravedad en rangos de edad más bajos.
Aunque los hallazgos no son del todo concluyentes, podrían permitir prever –y actuar en consecuencia ante– los casos de pacientes con una predisposición genética a sufrir una enfermedad de mayor gravedad y a requerir de ingreso en UCI, así como contribuir a conocer mejor la fisiopatología de la enfermedad. Podría plantearse, incluso, que un estudio precoz de las variantes genéticas de las regiones de los cromosomas 3 y 9 tendría cierto valor pronóstico, y considerarse como un factor de riesgo adicional a otras enfermedades crónicas (como diabetes, hipertensión u obesidad).
Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P et al. Genomewide Association Study of Severe Covid-19 with Respiratory. N Engl J Med. 2020; NEJMoa2020283. DOI: 10.1056/NEJMoa2020283.

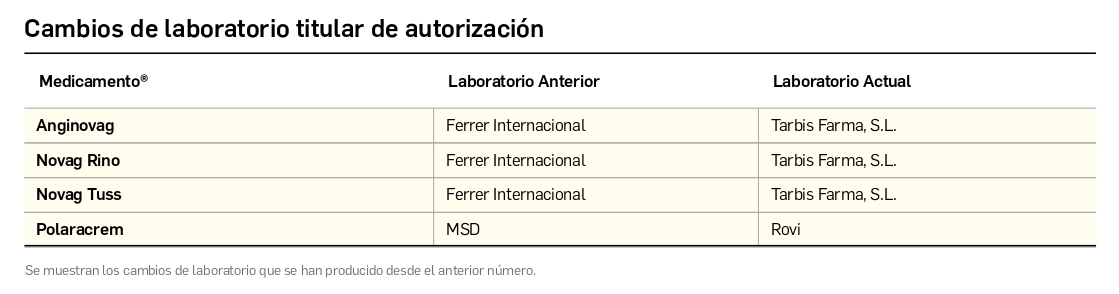
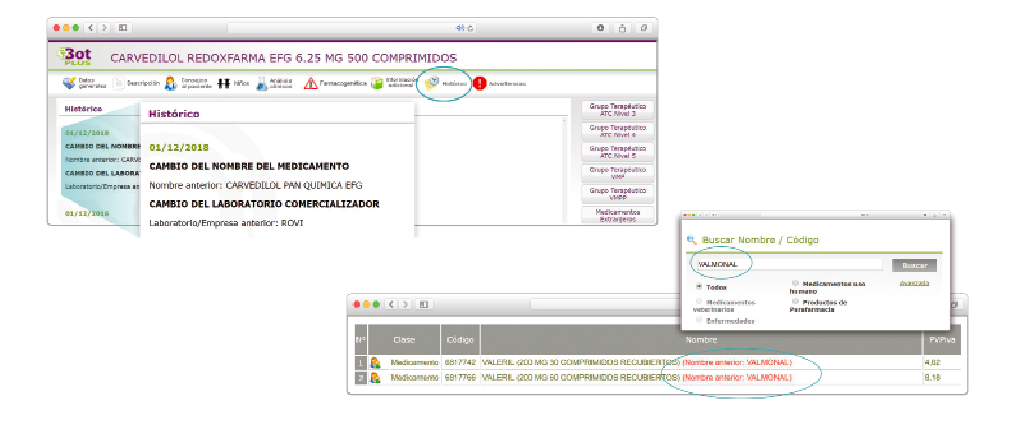

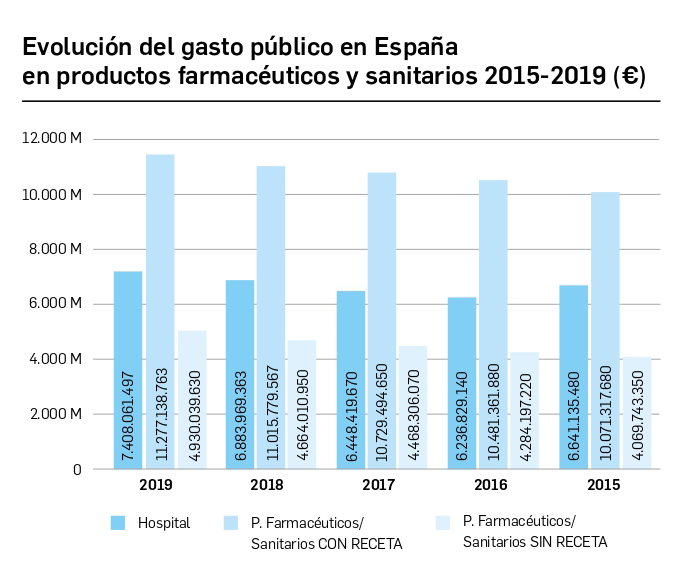
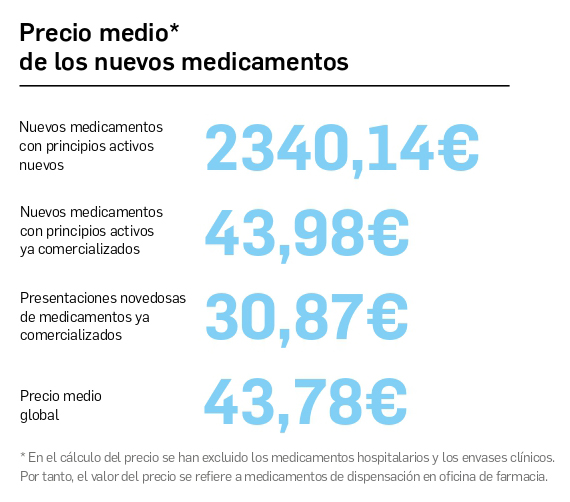
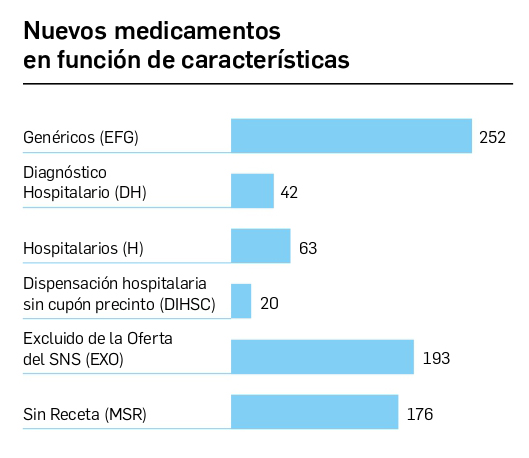


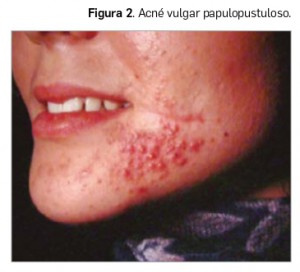
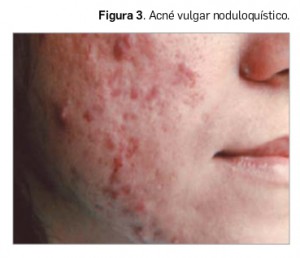
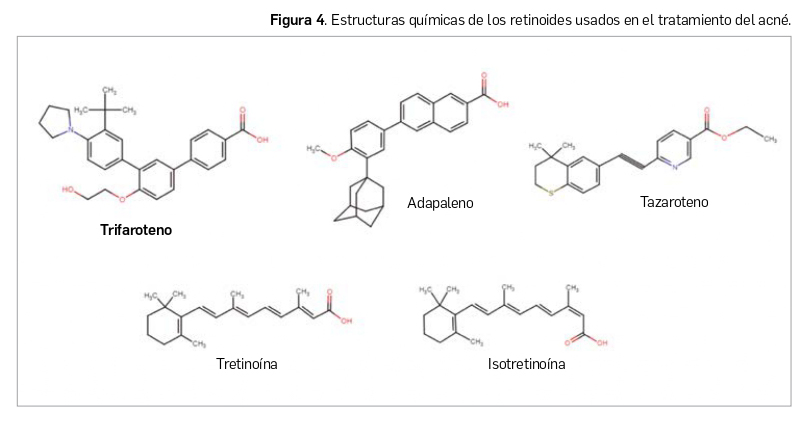
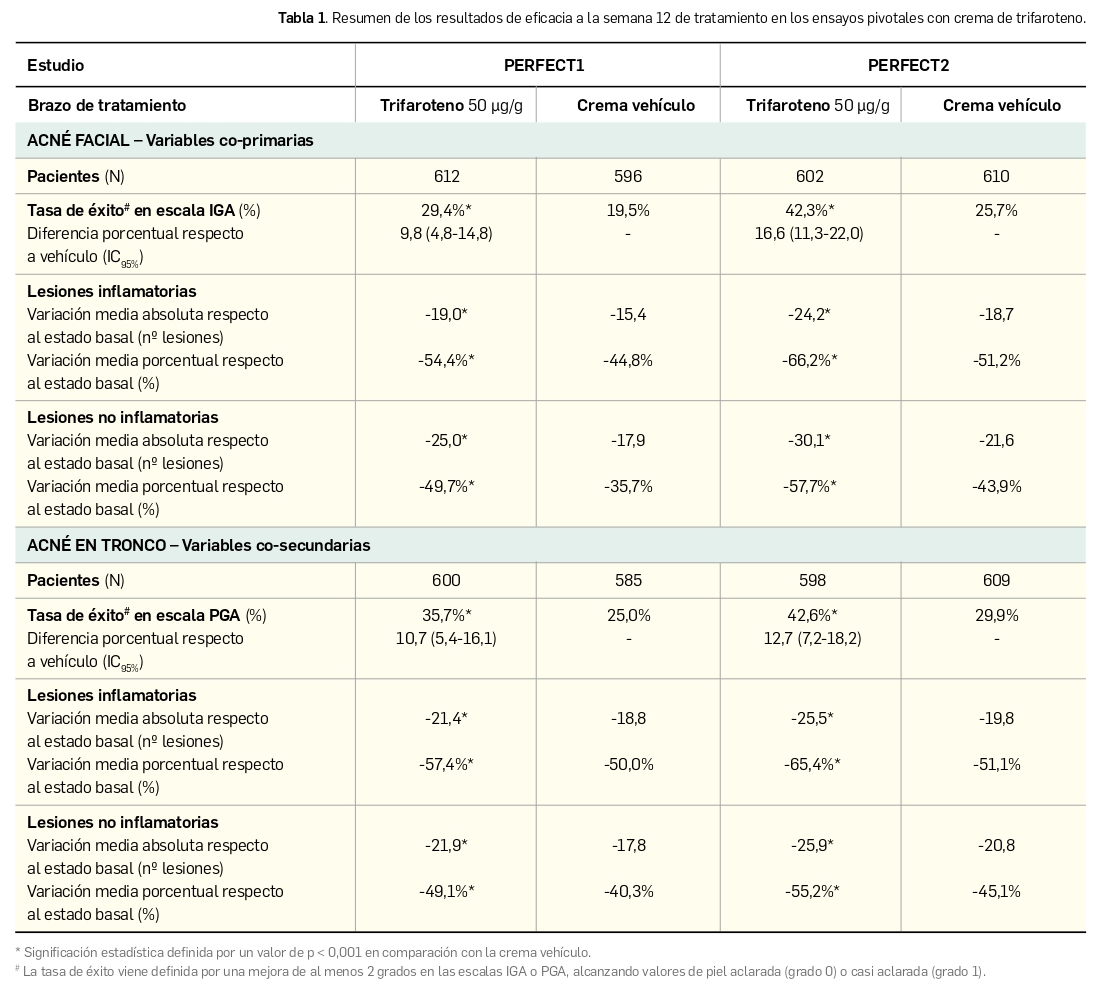




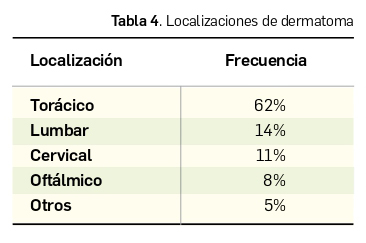
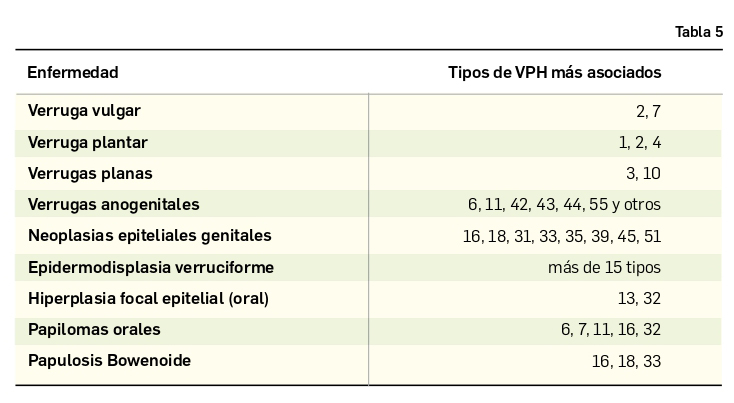
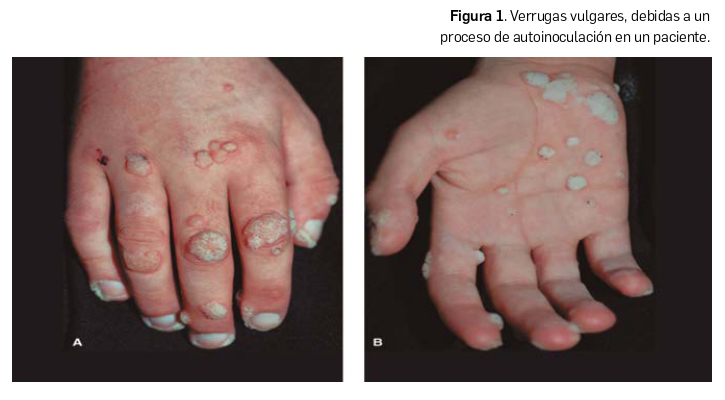
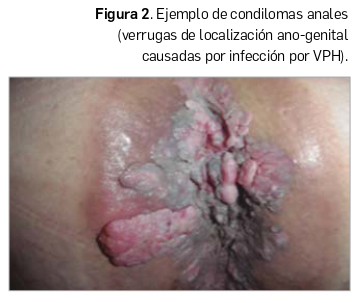
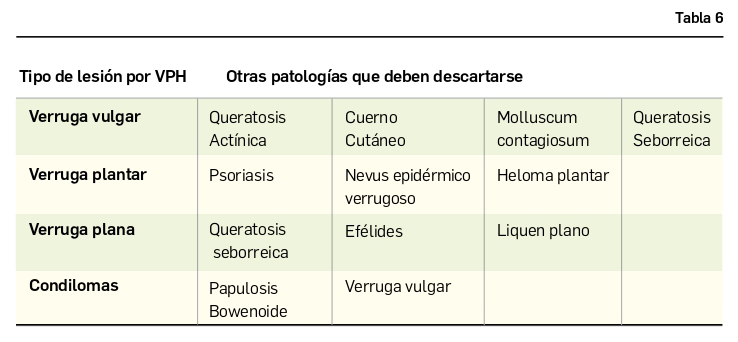
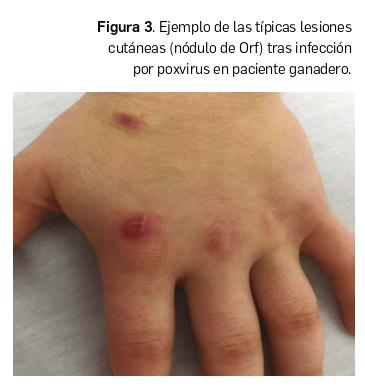


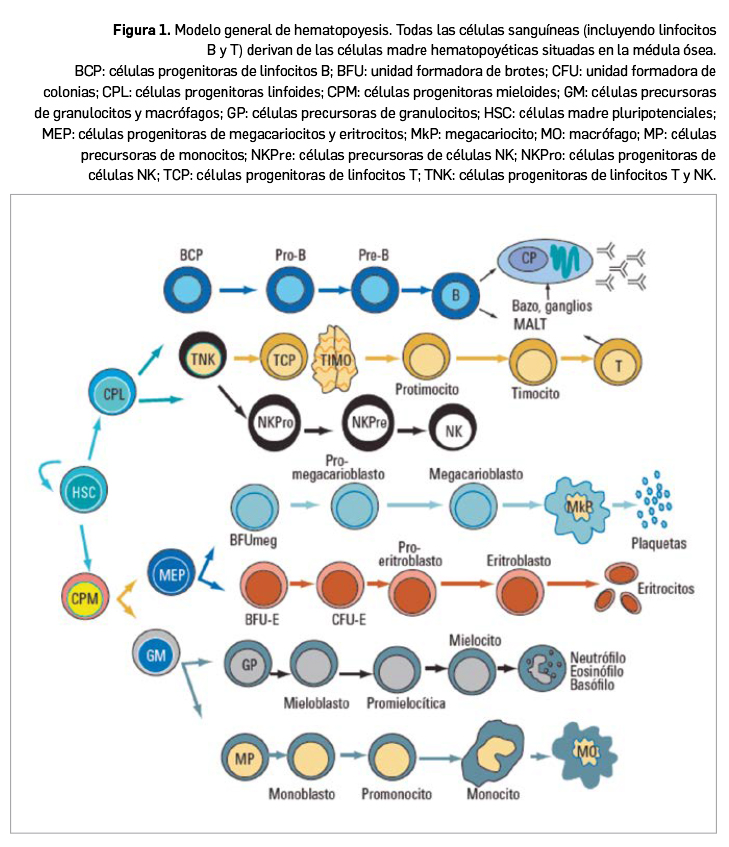
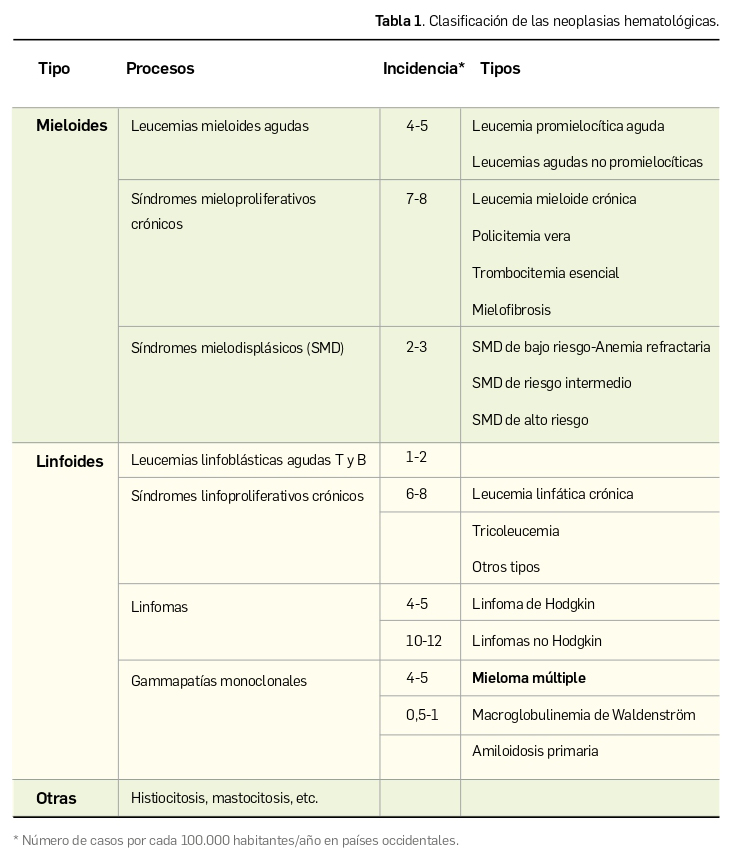
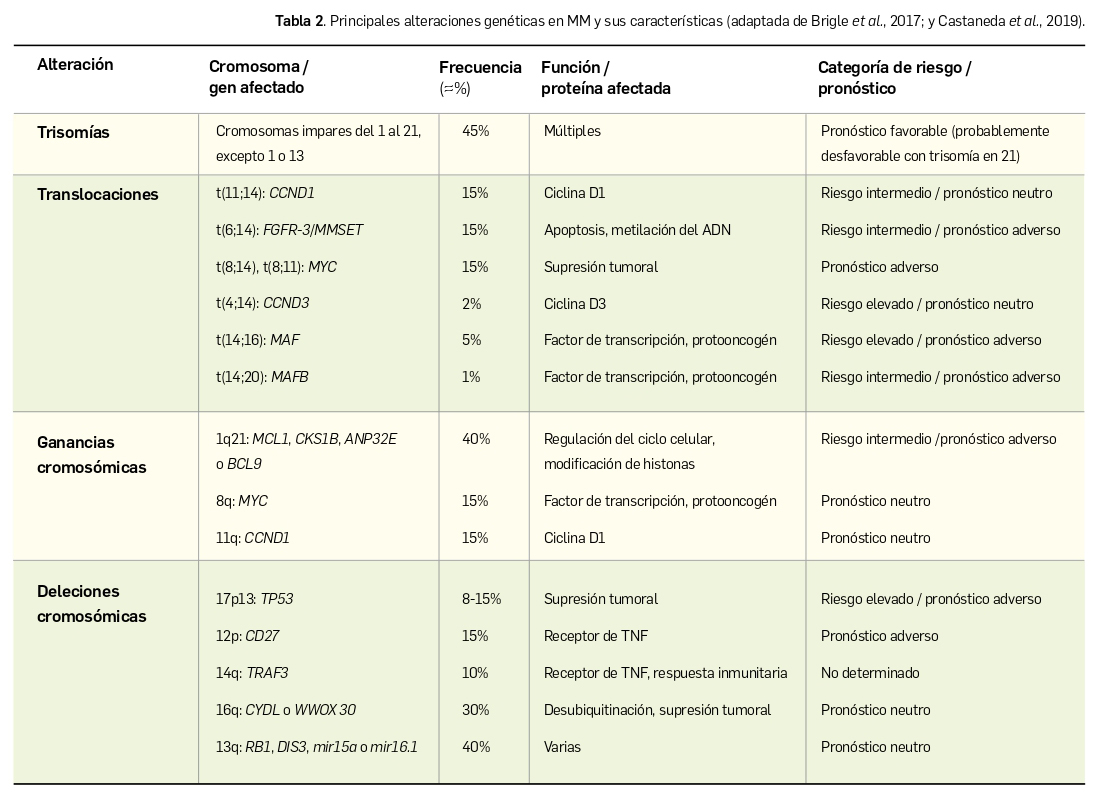

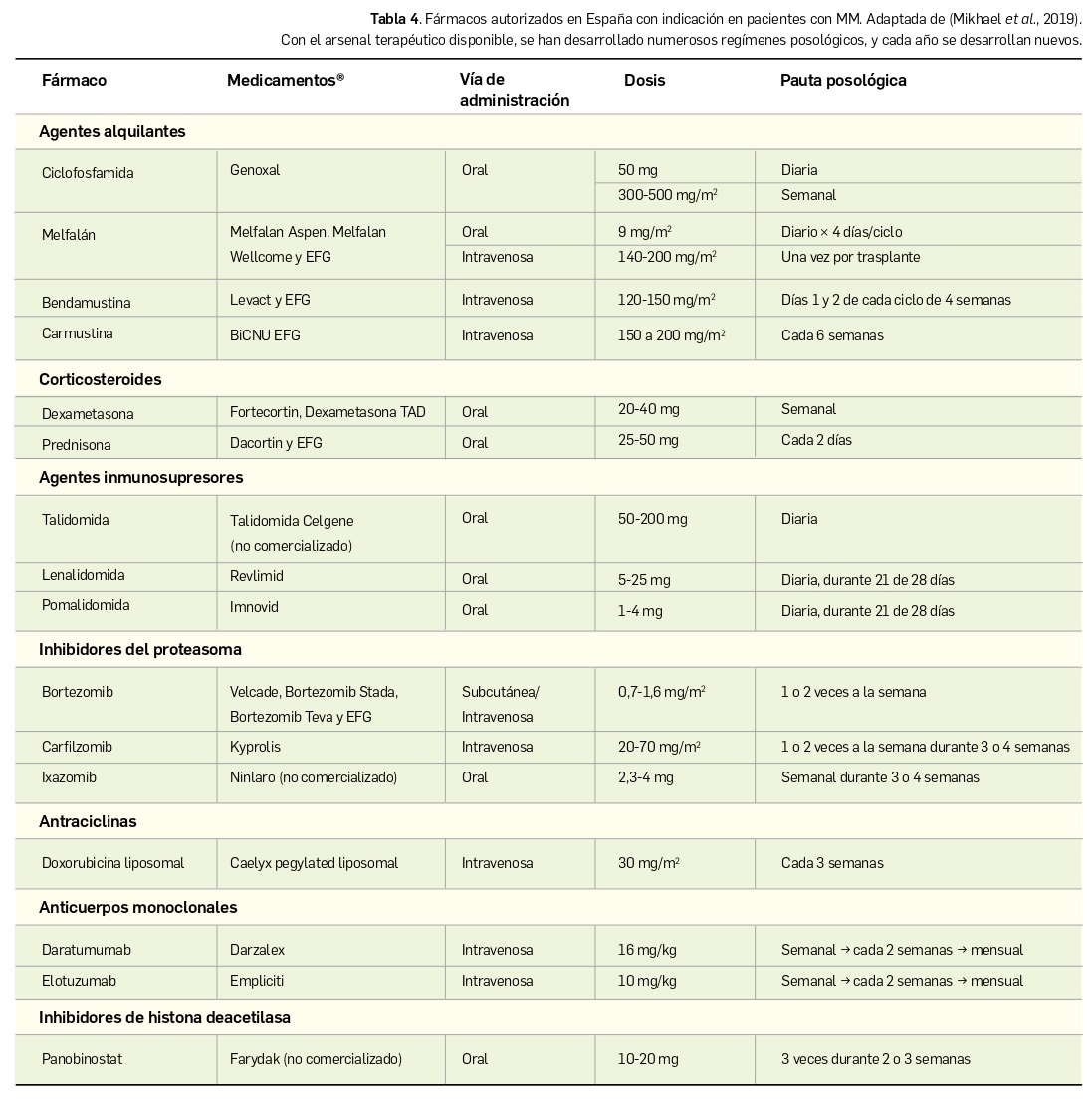
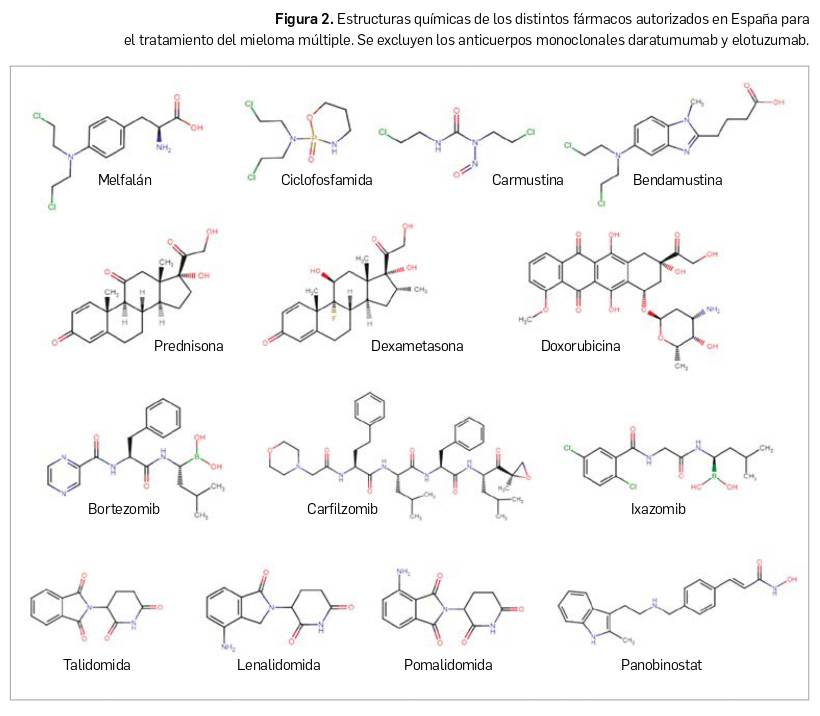
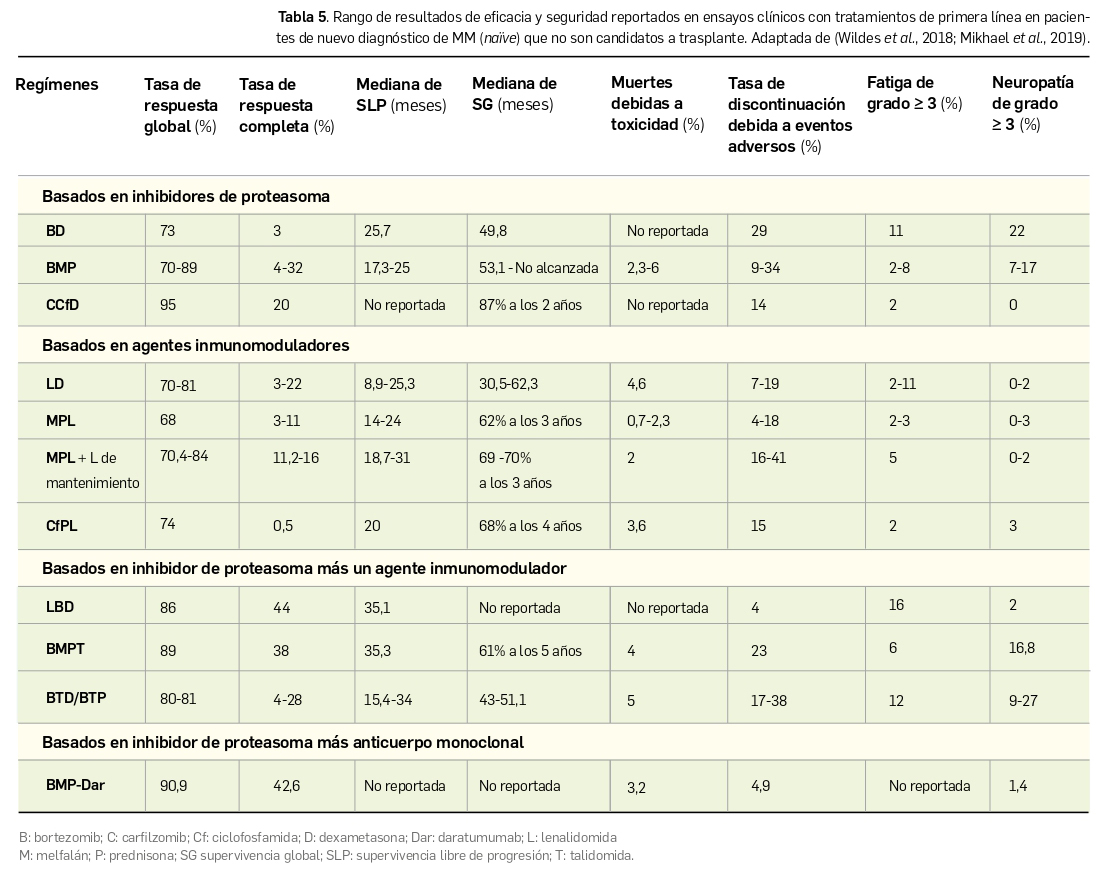
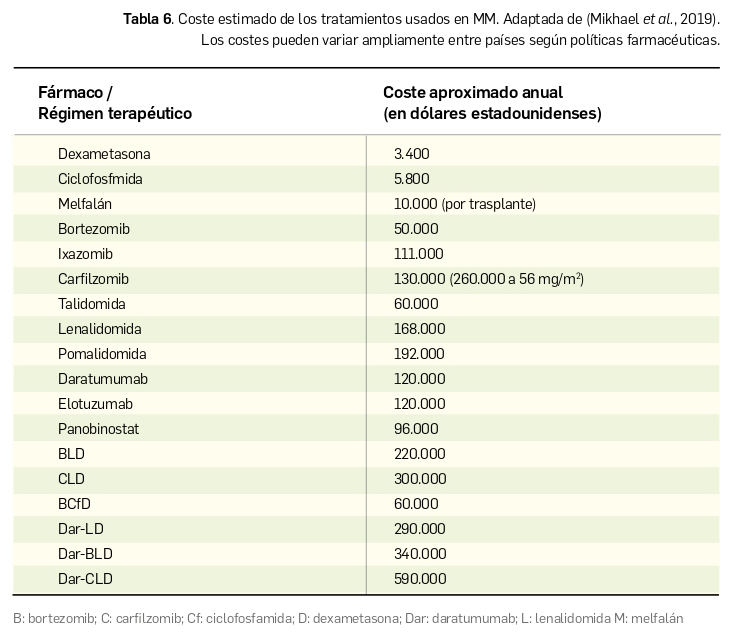

{kind=link}
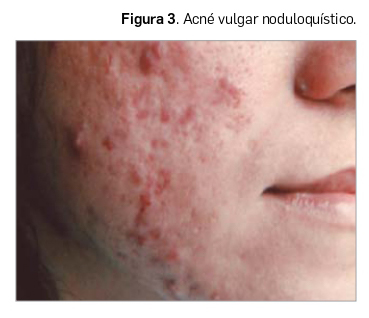{kind=link}