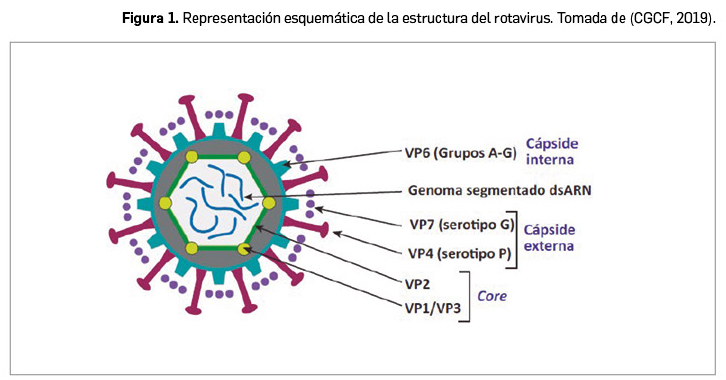
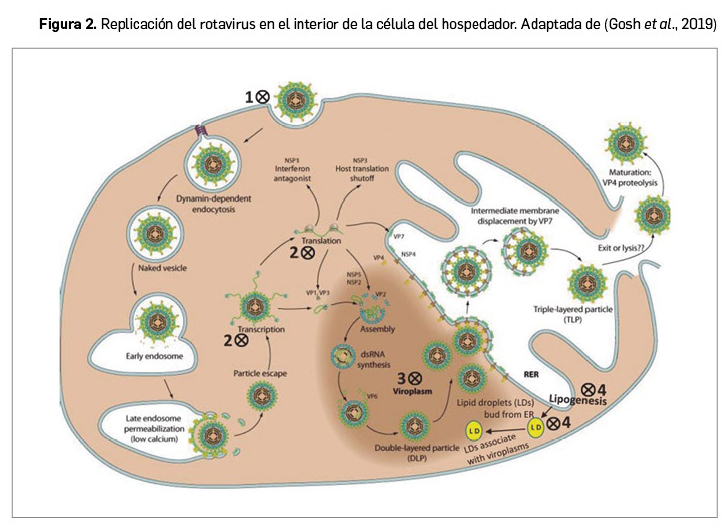
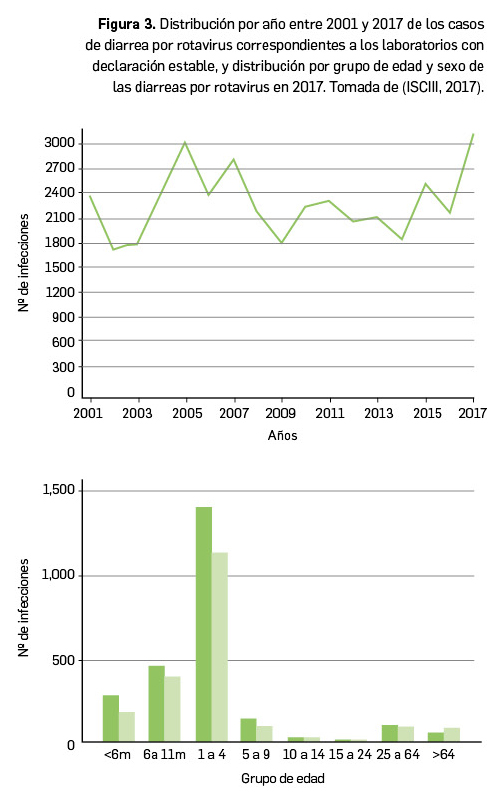
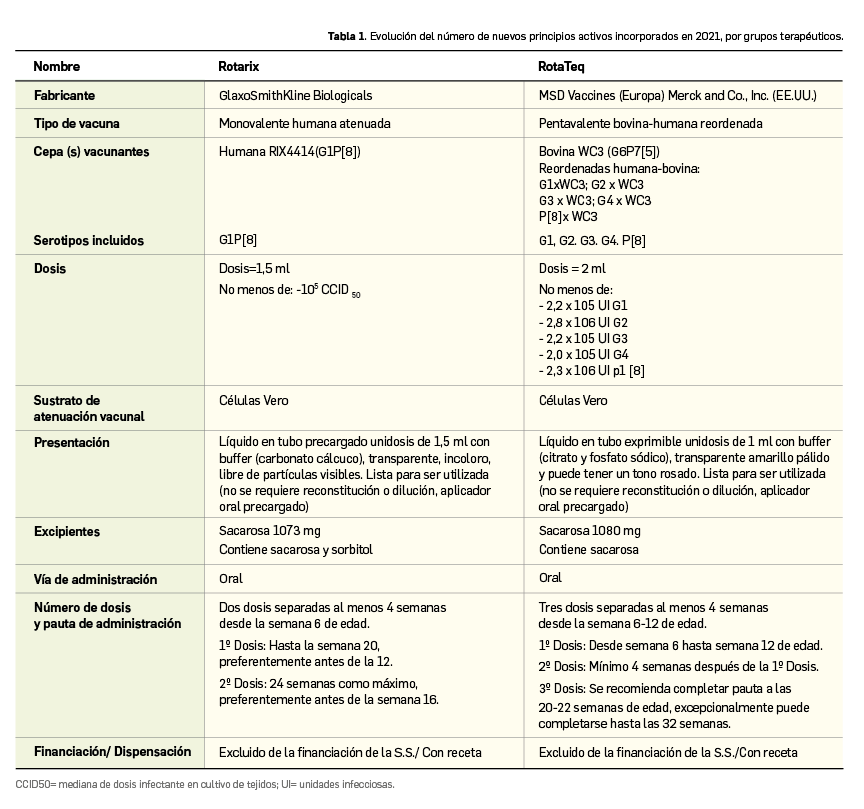
Resumen
Como viene siendo habitual, inauguramos el primer número anual de Panorama Actual del Medicamento con un resumen de todos los medicamentos con nuevos principios activos comercializados por primera vez en España en el último año. Comparativamente con sus predecesores, y a pesar de la imperante pandemia por COVID-19, el difícil 2021 ha sido un año bastante prolífico en cuanto a la incorporación al arsenal terapéutico de innovaciones farmacológicas, habiéndose comercializado un total de 35 nuevos principios activos, 24 más que en el año anterior; aproximadamente la mitad se enmarcan los grupos terapéuticos ATC L (11, terapia antineoplásica y agentes inmunomoduladores) y J (7, antiinfecciosos para uso sistémico). Continúa, pues, la tendencia del último lustro en que la mayor parte de los nuevos principios activos se dirigen al tratamiento de patologías oncológicas o de naturaleza autoinmune, si bien en este 2021 la autorización de medicamentos frente a la COVID-19 también ha hecho crecer en abundancia el grupo de los antiinfecciosos. En el último año, además, ha crecido en gran medida –hasta 12– el número de principios activos en medicamentos designados como huérfanos, desde los solo 2 en el año 2020.
A nivel de volumen de medicamentos, hay que destacar que por quinto año consecutivo se reduce el número de presentaciones de medicamentos comercializadas, pues en 2021 se han comercializado 1.136 nuevas presentaciones –tanto de nuevos principios activos como de los ya existentes– frente a las 1.468 que se han dado de baja. A finales del año, el mercado de medicamentos en España contaba con aproximadamente 18.000 formatos o presentaciones comerciales de medicamentos. En la última década se han incorporado 9.373 presentaciones, lo que supone un 52% del total, y han desaparecido 10.498, con un balance negativo de 1.125 formatos.
En cuanto al grado de innovación terapéutica, destaca sobremanera la comercialización y uso en España de los dos primeros medicamentos para la profilaxis farmacológica de la COVID-19 (Comirnaty® y Spikevax®, merecedores del Premio Panorama 2021), desarrollados y autorizados en menos de 1 año desde la identificación del SARS-CoV-2 como agente causal, gracias a procesos adaptativos pioneros en investigación clínica y de revisión de la evidencia por agencias reguladoras. Se trata de las dos primeras vacunas basadas en ARNm indicadas en humanos, una tecnología completamente novedosa que abre un campo farmacoterapéutico muy prometedor por la especificidad de los fármacos, donde ya hay candidatos que se están estudiando frente otras patologías humanas, incluidas las oncológicas. Con una eficacia y seguridad clínicas contrastadas, y confirmadas con su uso masivo posautorización, han conllevado un impacto enormemente positivo en la lucha frente a una pandemia que ha supuesto la mayor crisis sanitaria, social y económica en todo el mundo en décadas.
Las principales características fármaco-clínicas de todos los nuevos principios activos de 2021 se resumen en el presente artículo.
___________________________________________________________________________________________
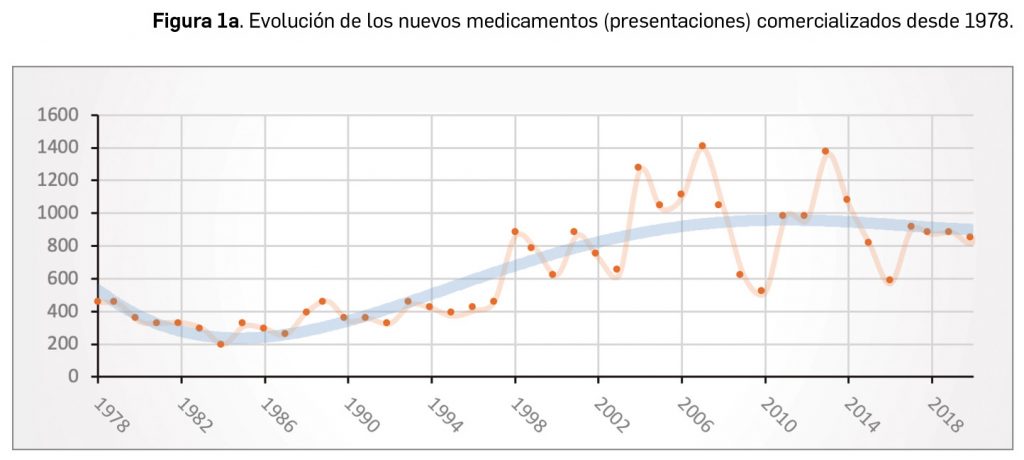
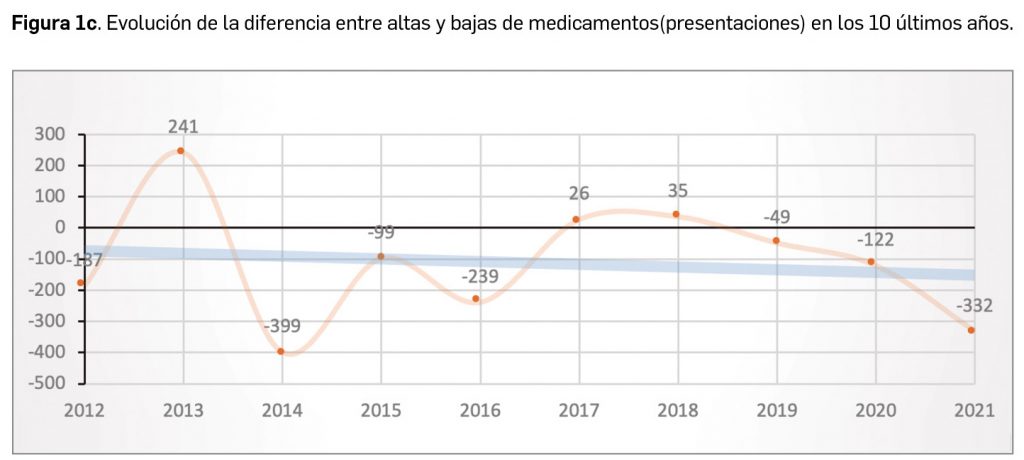
Durante el año 2021, se registraron en España 1.136 nuevas presentaciones comerciales o formatos de medicamentos, tal y como se recoge en la Tabla 1, incluyendo el 91% de ellas principios activos previamente comercializados. El dato se alinea con la tendencia mostrada en los periodos analizados, en los últimos 43 años (Figura 1a) y en los últimos 10 años (Figura 1b).
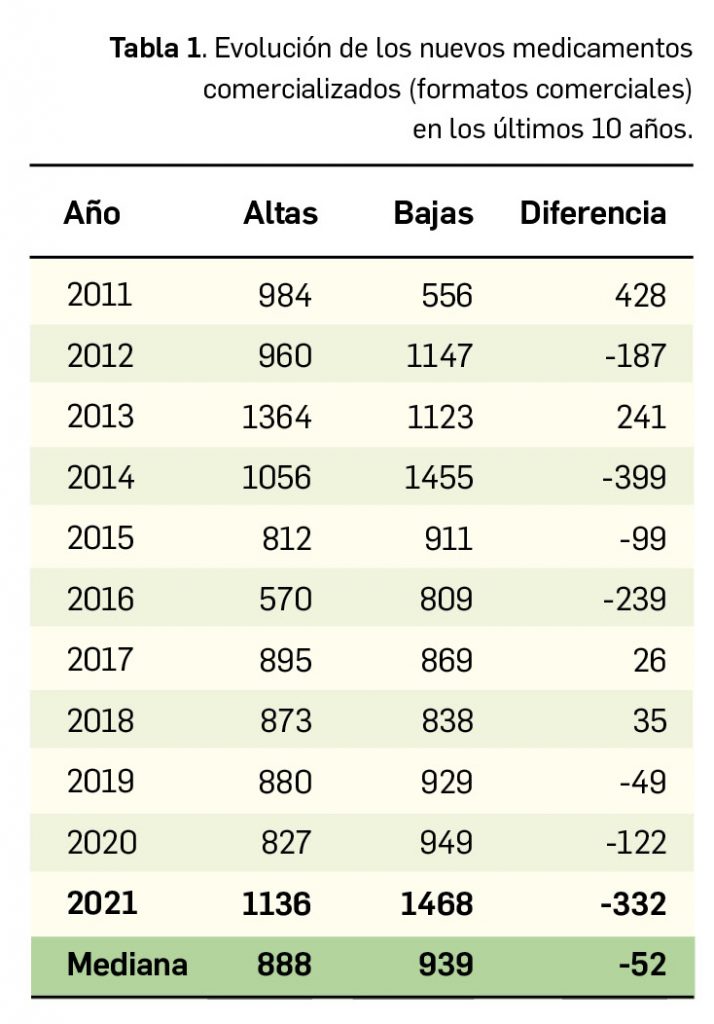
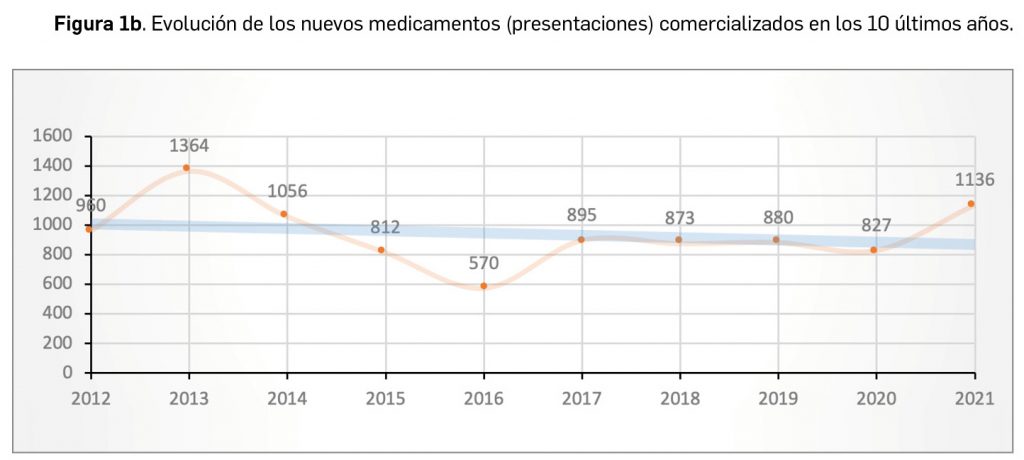
Cabe destacar, por ejemplo, que un total de 591 presentaciones corresponden a medicamentos genéricos, lo cual representa el 52,0% del total de nuevas presentaciones. A fecha de 31 de diciembre de 2021, se encontraban en situación de comercialización un total de 18.141 presentaciones de medicamentos. Por otro lado, durante los últimos 10 años se han incorporado al mercado 9.373 presentaciones, lo que supone el 51,7% del total disponible actualmente; no obstante, también desaparecieron 10.498 presentaciones, lo cual se traduce en un descenso neto de 1.125 formatos en ese periodo. La tendencia a la renovación viene determinada por el incremento del número de bajas respecto a periodos anteriores y la paulatina estabilización del número de altas en los últimos 5 años (solo interrumpida en este 2021), en los que ha habido un total de 1.125 más bajas que altas (Figuras 1b y 1c).
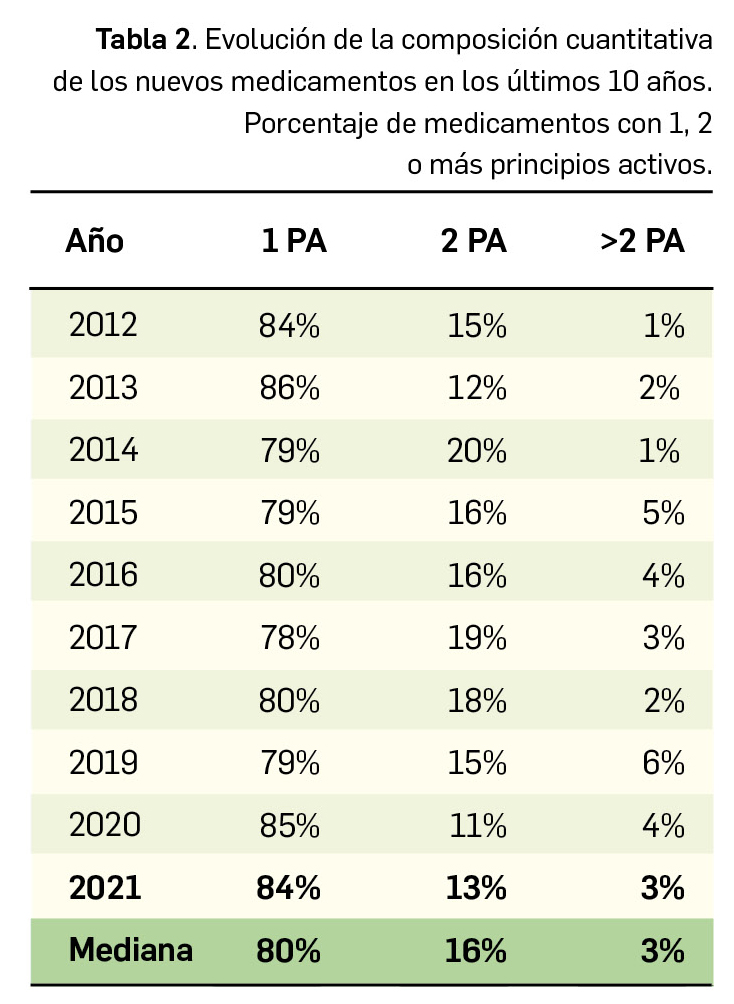
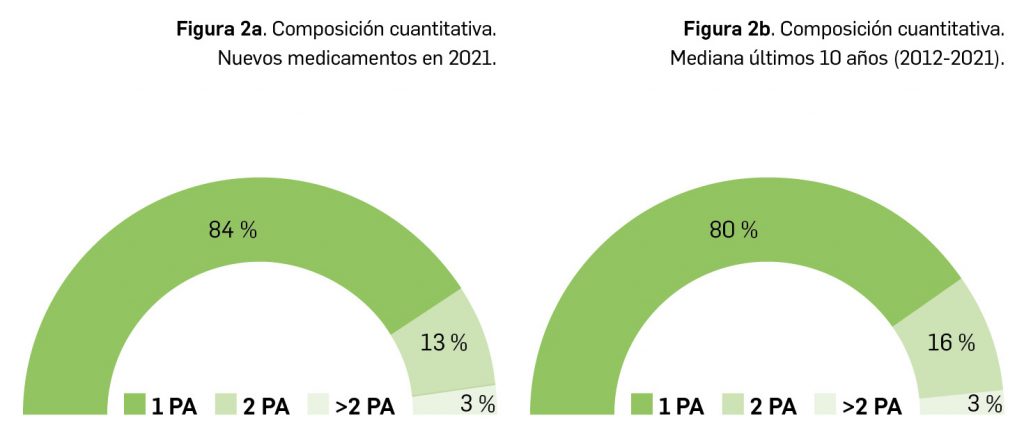
En lo relativo a la composición cuantitativa de los nuevos medicamentos comercializados en 2021, el 84% de estos fueron monocomponente, un 13% contenían dos principios activos y el restante 3% eran medicamentos multicomponente (Figura 2a). En este sentido, parece evidente que se mantiene la tendencia hacia los medicamentos monocomponente, que suponen el 80% de los nuevos medicamentos comercializados en los últimos 10 años (Tabla 2 y Figura 2b).
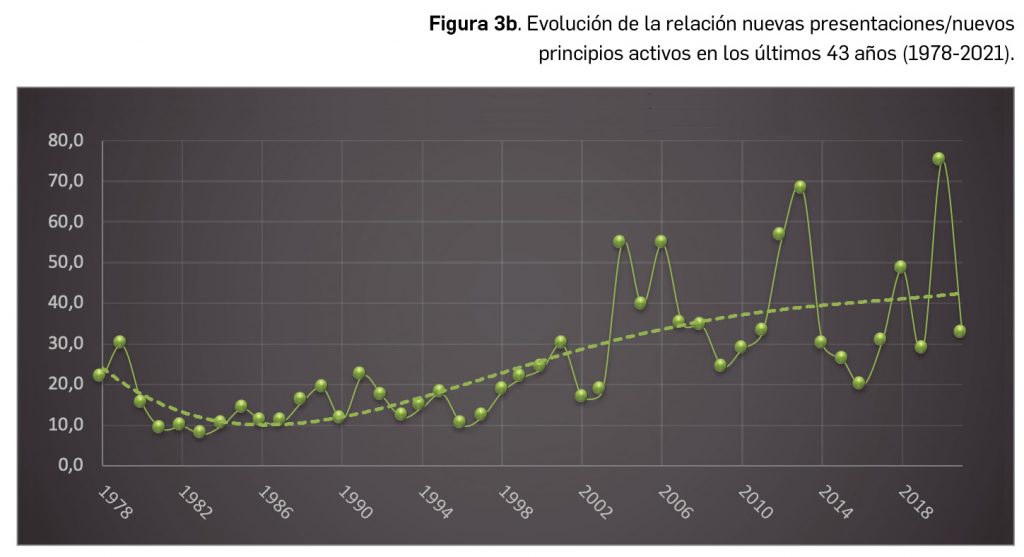
Por lo que se refiere a los nuevos principios activos comercializados en 2021 en nuestro país, han sido un total de 35 –de los cuales hasta 12 han sido incluidos en medicamentos designados como como huérfanos (un 34%)– (Tabla 3), 24 más que el año pasado, lo que representa un número ligeramente superior a la mediana de 29 nuevos principios activos/año correspondiente a la década 2012-2021 (el promedio de nuevos principios activos/año se sitúa en 25,6). Teniendo en cuenta que se han comercializado un total de 1.136 presentaciones de medicamentos en 2021, se obtiene un ratio de 32,5 presentaciones nuevas de medicamentos por cada nuevo principio activo comercializado1, similar superior a la mediana de la última década (30,6). En este sentido, cabe destacar que el 91% de los nuevos medicamentos comercializados durante 2021 incluyeron principios activos autorizados y comercializados en años previos; de ellos, un 3% son presentaciones novedosas de medicamentos ya comercializados previamente, entendiendo como tal aquellas que suponen una innovación en forma farmacéutica y/o vía de administración.
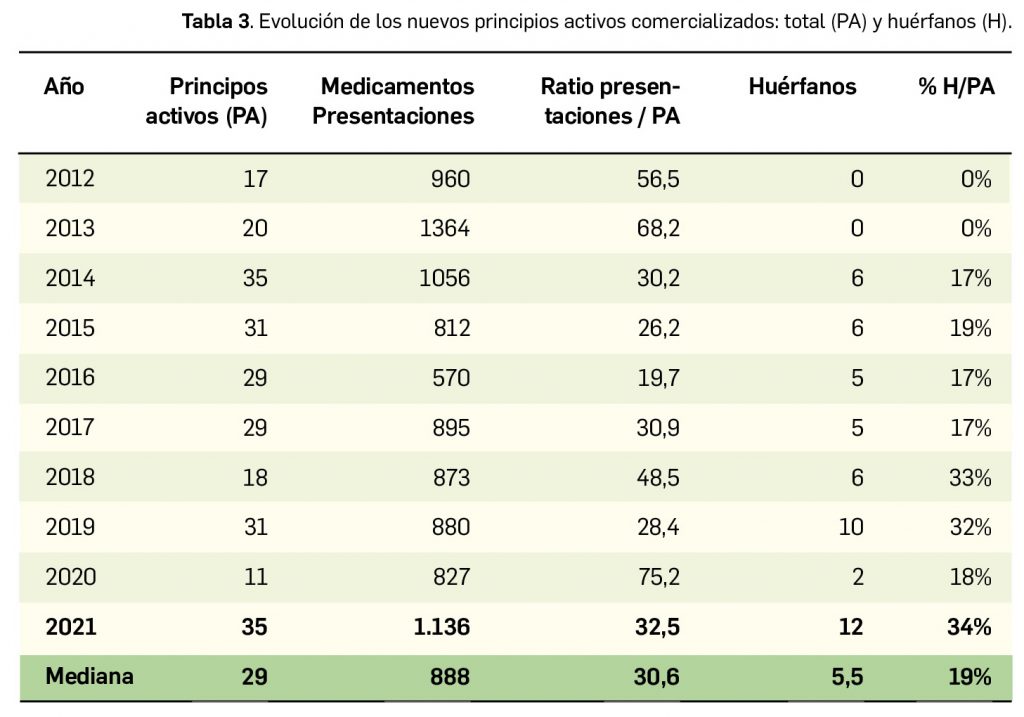
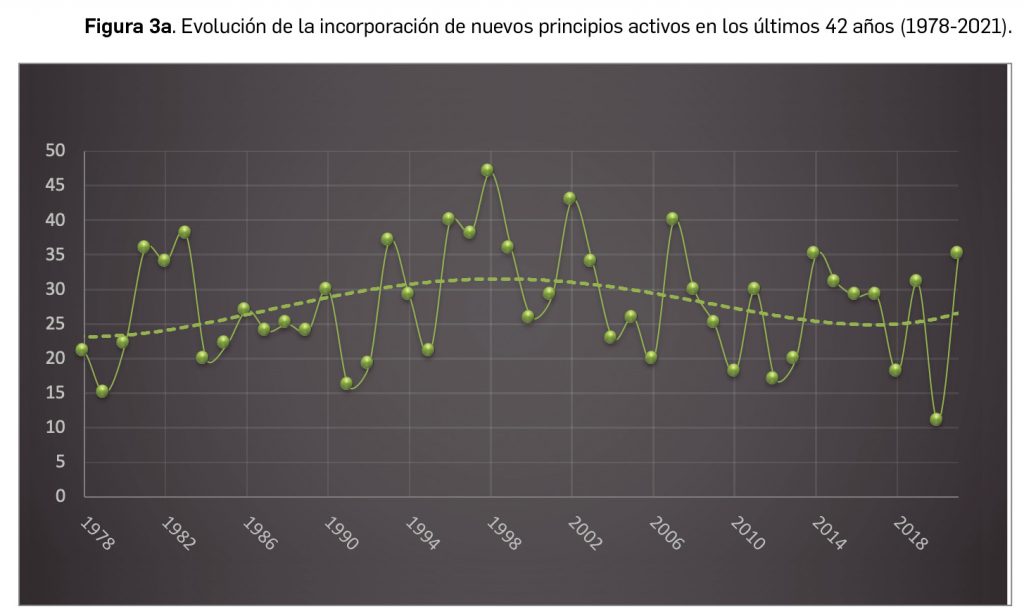
A grandes rasgos, las tendencias de la incorporación de nuevos principios activos en los últimos 43 años (1978-2021) (Figura 3a) y de la relación nuevas presentaciones/nuevos principios activos (Figura 3b) son moderadamente fluctuantes2. En los últimos 10 años se aprecia una ligera estabilización en la incorporación de nuevos principios activos en el mercado español (Figura 3c), que fue interrumpida por el año 2020 (en el que se produjo la menor comercialización de principios activos nuevos desde que se tienen registros en PAM), pero se ha recuperado en 2021. Este año la relación global nuevas presentaciones/nuevos principios activos (32,5) ha vuelto a valores muy cercanos a la mediana de la última década (30,6) .
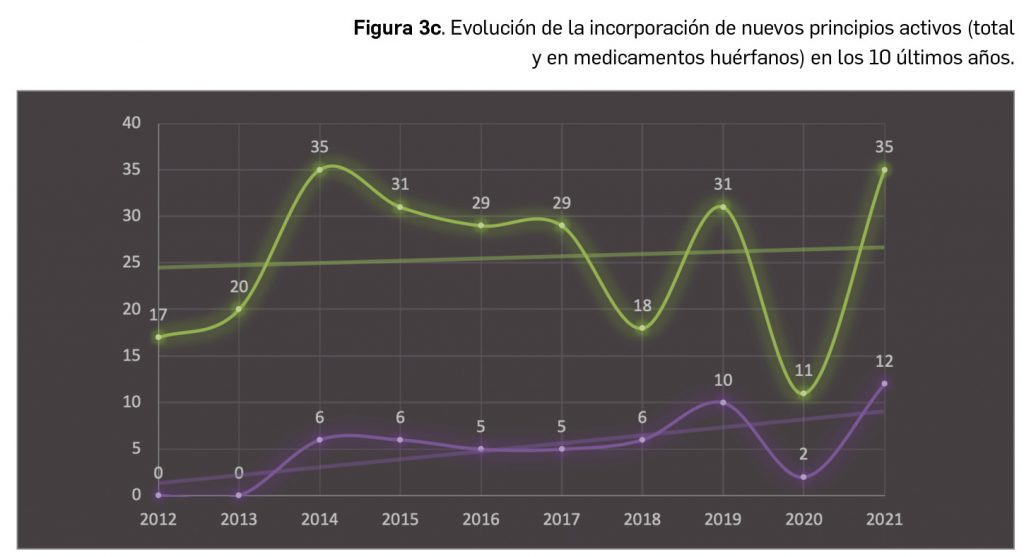
Desde el año 2002, se han comercializado en España 105 nuevos principios activos como medicamentos huérfanos3, lo que supone un 19% de los nuevos principios activos incorporados en ese periodo (un total de 545). La tendencia fue ligeramente ascendente hasta 2019 gracias a los 32 nuevos medicamentos huérfanos incorporados en los 5 años previos (2015-2019), pero también se vio interrumpida en 2020 por los solo 2 nuevos principios activos incluidos en medicamentos que han sido designados como huérfanos. Así pues, se ha recuperado con fuerza en el pasado 2021 la comercialización de medicamentos huérfanos. En cualquier caso, parece mantenerse cierta proporcionalidad entre el número total de nuevos principios activos y el de los incluidos específicamente en medicamentos huérfanos en cada año, lo que queda reflejado en el paralelismo entre ambas líneas de tendencia (Figura 3c).
Por otro lado, resulta reseñable que en el año 2021 se han comercializado en España 6 nuevos medicamentos biosimilares: dos del principio activo bevacizumab, uno de adalimumab, uno de trastuzumab, uno de pegfilgrastim y uno de teriparatida.
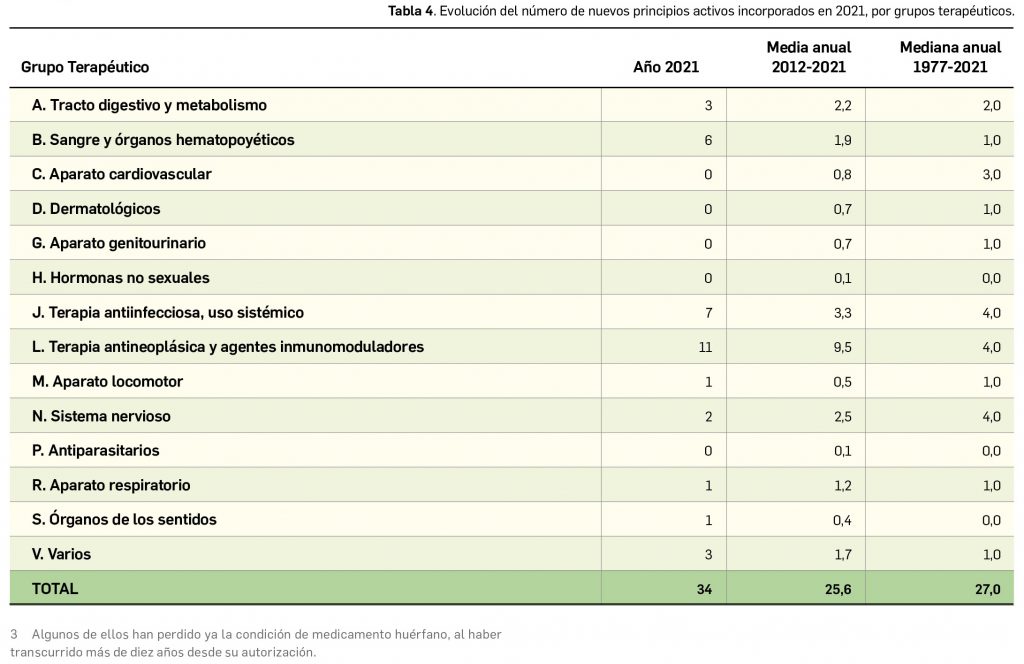
Si se considera la clasificación terapéutica ATC de los nuevos principios activos comercializados en 2021, se han incorporado principios activos a 9 de los 14 grupos terapéuticos existentes. El grupo con mayor número de nuevos principios activos durante el año ha sido, como ya venía ocurriendo en los últimos años, el grupo L (Terapia antineoplásica y agentes inmunomoduladores), con un total de 11. También cabe destacar, en un año marcado por la COVID-19, la comercialización de 7 nuevos medicamentos enmarcados en el grupo J (antiinfecciosos para uso sistémico).
A modo de resumen, desde el año 1977, en que apareció por vez primera Panorama Actual del Medicamento, se han incorporado un total de 1.242 nuevos principios activos al mercado farmacéutico español, con independencia de su clasificación terapéutica ATC.
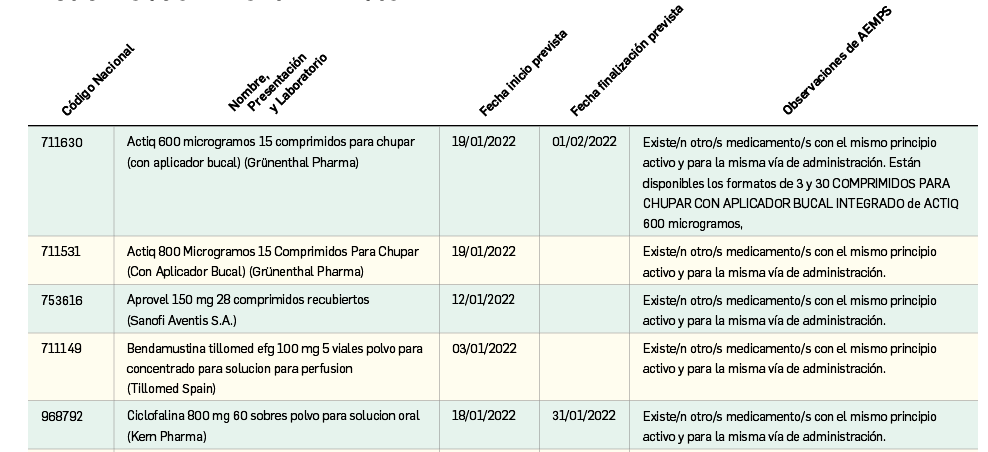
En la siguiente tabla (Tabla 5) se muestran los nombres de los nuevos principios activos comercializados durante el año 2021, junto a los nombres de los medicamentos en que se incluyen, su grupo terapéutico ATC e indicación principal. Seguidamente, se resumen las principales características fármaco-clínicas de cada uno de ellos, clasificados por grupos terapéuticos, en base a la información disponible en el momento de su primera comercialización en España. Para algunos de los medicamentos consignados en la tabla no se incluyen sus resúmenes, los cuales aparecerán en los próximos números de Panorama Actual del Medicamento.
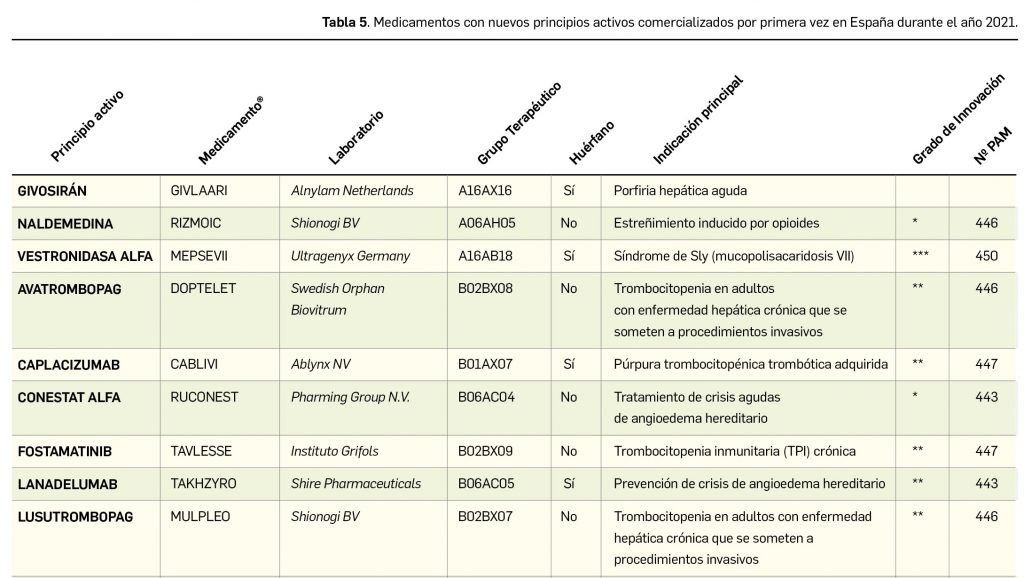
Continúa la tabla aquí
Tracto alimentario y metabolismo
Estreñimiento inducido por opioides
Naldemedina ▼RIZMOIC® (Shionogi BV) PAM 446
Naldemedina es un nuevo antagonista –derivado de naltrexona– de los receptores opioides mu, delta y kappa presentes en los tejidos periféricos, que produce un efecto laxante por su antagonismo en los receptores mu del sistema nervioso entérico del tubo digestivo. Dada su escasa capacidad de atravesar la barrera hematoencefálica, reduce los efectos astringentes de los opioides sin revertir su efecto analgésico mediado por el sistema nervioso central. El medicamento ha sido autorizado para el tratamiento –por vía oral– del estreñimiento inducido por opioides (EIO) en pacientes adultos que han recibido previamente tratamiento con un laxante.
En dos estudios con pacientes no oncológicos con EIO (COMPOSE-1 y -2) y sin uso concomitante de otros laxantes, un tratamiento diario con naldemedina durante 12 semanas aumentó en un 13-19% la tasa de respondedores en comparación con placebo; las variables secundarias confirmaron que, además de una mejora en la frecuencia semanal de deposiciones espontáneas en general, induce un incremento en las deposiciones completas y en aquellas sin esfuerzo defecatorio. El beneficio es significativo ya desde la primera semana y se mantiene posteriormente. Un tercer estudio (COMPOSE-3) corroboró la superioridad de naldemedina en tratamientos de hasta 1 año de duración, con uso concurrente o no al de laxantes, pues permite casi duplicar el promedio de deposiciones semanales, con una diferencia reseñable frente a placebo. Adicionalmente, en pacientes con dolor crónico por cáncer y EIO, los datos agrupados de dos estudios (el pivotal COMPOSE-4 y otro de búsqueda de dosis) evidenciaron una mejora sustancial en la tasa de respondedores con tratamientos de 2 semanas con naldemedina (71-78%): el incremento se situó entre el 37-40% en comparación con el placebo. La eficacia del nuevo fármaco fue consistente en todos los subgrupos de pacientes, con independencia de su respuesta –inadecuada o no– a otros laxantes.
En términos de seguridad, naldemedina parece bien tolerado en todas las poblaciones de pacientes, quizás en mayor medida que otros antagonistas opioides disponibles en la misma indicación (metilnaltrexona, naloxegol), pues no ha provocado perforaciones intestinales. Su perfil toxicológico se caracteriza por efectos adversos gastrointestinales posiblemente debidos a su mecanismo de acción, siendo los más frecuentes el dolor abdominal, la diarrea (más incidente en pacientes oncológicos), las náuseas y los vómitos. La mayoría son de gravedad leve a moderada, y la tasa de interrupción del tratamiento es baja (3-4,5%). Con una frecuencia muy baja (< 1%), naldemedina podría producir algún caso de síndrome de abstinencia a opioides con manifestaciones extradigestivas, independiente de la terapia opioide de mantenimiento.
Así pues, el fármaco ha demostrado ser clínicamente superior a placebo –en base a variables referentes a la frecuencia de defecaciones– en el tratamiento del estreñimiento inducido por opioides en pacientes con dolor crónico de origen oncológico y no oncológico, con independencia de la respuesta previa a otros laxantes. Parece que su eficacia es mayor en pacientes con cáncer. A falta de estudios comparativos, el IPT establece que naldemedina podría considerarse una opción de tratamiento laxante comparable a naloxegol y metilnaltrexona, con los que comparte mecanismo de acción, por lo que no parece incorporar un grado sustancial de innovación terapéutica. No obstante, frente a naloxegol, naldemedina aporta la ventaja potencial de que puede usarse con otros laxantes, y frente a metilnaltrexona, sobresale su vía de administración oral (más sencilla que la vía subcutánea) y el menor coste del nuevo medicamento.
Sangre y órganos hematopoyéticos
Púrpura trombocitopénica trombótica adquirida
Caplacizumab ▼CABLIVI® (Ablynx NV) PAM 447
Caplacizumab es un nanoanticuerpo bivalente humanizado que se une con especificidad y afinidad al dominio A1 del factor de von Willebrand (FvW) e inhibe su interacción con las plaquetas, impidiendo la adhesión plaquetaria que median sus multímeros de alto peso molecular. Por abordar una característica fisiopatológica principal de la enfermedad, el medicamento, designado como huérfano, ha sido autorizado para el tratamiento –por vía subcutánea e intravenosa– de adultos y adolescentes a partir de 12 años y 40 kg de peso que presentan un episodio de púrpura trombocitopénica trombótica adquirida (PTTa), junto con intercambio plasmático e inmunosupresión.
Su aprobación se sustentó en los datos clínicos de un ensayo pivotal aleatorizado de fase 3, en el que 144 pacientes adultos con episodios agudos de PTTa recibieron caplacizumab o placebo concomitantemente a la terapia estándar combinada de recambio plasmático e inmunosupresión farmacológica. Un tratamiento con la pauta diaria autorizada del fármaco acortó de forma discreta el tiempo hasta la normalización del recuento plaquetario, en un día y medio sobre el seguimiento completo de 20 días; frente a placebo, los pacientes tenían una probabilidad un 55% mayor de lograr respuesta clínica en cualquier momento. Si bien la relevancia clínica de esos resultados podría cuestionarse, las variables secundarias confirmaron la superioridad de caplacizumab sobre placebo: redujo en un 74% la proporción de pacientes que cumplía una variable combinada de muerte, recurrencias o evento trombótico mayor por la enfermedad; disminuyó la tasa de recurrencias en un 67%; y redujo el número medio de días de recambio plasmático, el volumen medio de plasma usado y la duración de las estancias hospitalarias y en las UCI. Pero la eficacia parece modesta, dado que no se vieron diferencias en el número de eventos tromboembólicos mayores y no se pudo demostrar un beneficio en términos de refractariedad al tratamiento ni en la normalización de biomarcadores de daño tisular.
El perfil toxicológico de caplacizumab es aceptable, manejable clínicamente y consistente en todos los subgrupos de pacientes. Muestra una buena tolerabilidad: la tasa de eventos adversos graves fue menor que con placebo, y menos de la mitad de ellos se consideraron como posiblemente relacionados el fármaco (14% vs. 6% en el grupo placebo). El principal problema de seguridad es el aumento del riesgo de hemorragias, generalmente leves-moderadas y mayoritarias a nivel de piel y mucosas. Los acontecimientos adversos más frecuentes fueron epistaxis o sangrado nasal (18%) y hematomas, junto con cefalea (21%), hemorragia gingival y petequias. Se debe suspender el tratamiento en caso de hemorragia activa o la realización de cirugía, y monitorizar a pacientes que usen fármacos anticoagulantes o antiagregantes. La práctica totalidad de eventos adversos relacionados con el sistema inmunitario (49% vs. 32% con placebo) fueron leves o moderados.
Dado que el actual tratamiento de primera línea de los episodios agudos de PTTa –inicio rápido de recambio plasmático junto con terapia inmunosupresora– es ineficaz en hasta un 20% de los pacientes, caplacizumab puede representar una alternativa terapéutica en aquellos con respuesta insuficiente al tratamiento estándar o en quienes la enfermedad recurra. Incorpora un nivel reseñable de innovación por tratarse del primer medicamento específicamente autorizado para tratar la PTTa y en abordar su fisiopatología, inaugurando una vía terapéutica y confirmando el potencial terapéutico que tienen los nanoanticuerpos derivados de camélidos. No obstante, no es un tratamiento curativo (no corrige el origen de la enfermedad: el déficit de actividad metaloproteinasa de ADAMTS13) y el beneficio clínico que aporta parece moderado. La necesidad de mantener la terapia estándar durante el tratamiento limita su grado de innovación.
Trombocitopenia en adultos con hepatopatía crónica
Avatrombopag ▼DOPTELET® (Swedish Orphan Biov.) PAM 446
Lusutrombopag ▼MULPLEO® (Shionogi BV) PAM 446
Se trata de dos moléculas pequeñas activas por vía oral que actúan como agonistas del receptor de la trombopoyetina (TPO) expresado en megacariocitos humanos y en las células progenitoras de médula ósea: estimulan su proliferación y diferenciación y, en consecuencia, provocan un aumento de la producción de plaquetas (trombopoyesis), sumando su efecto al de la TPO endógena al no competir por su unión al receptor. Los medicamentos con dichos principios activos han sido autorizados para el tratamiento de la trombocitopenia grave en adultos con hepatopatías crónicas (HC) que tengan programada una intervención invasiva. Avatrombopag también ha recibido aprobación para tratar la trombocitopenia inmunitaria primaria crónica (TIPC) en pacientes adultos que no responden a otros tratamientos (por ejemplo, corticosteroides, inmunoglobulinas).
En dos estudios pivotales de fase 3 casi idénticos con pacientes con HC y trombocitopenia grave, un tratamiento de 5 días con avatrombopag indujo un aumento estadísticamente significativo, en comparación con placebo, en el porcentaje de pacientes que no requirió una transfusión de plaquetas ni tratamiento de rescate por hemorragias hasta 7 días después de la intervención invasiva programada (66-88% vs. 23-38% con placebo); además, aumentó notablemente la proporción de pacientes con recuento plaquetario de ≥ 50.000 plaquetas/μl el día de la intervención. Igualmente, un tratamiento de hasta 7 días con lusutrombopag demostró en dos estudios pivotales de similar diseño (y población de pacientes con características parecidas) que es superior a placebo: casi triplicó la proporción de pacientes que no necesitaba transfusión de plaquetas ni otro tratamiento de rescate hasta 7 días después del procedimiento por tener niveles de > 50.000 plaquetas/μl (68% vs. 24%); otras variables secundarias relacionadas –porcentaje de sujetos que no necesitó transfusión desde el día 1 hasta el 35 y de pacientes respondedores– también favorecieron al tratamiento experimental. La eficacia de ambos fármacos es consistente en todos los subgrupos de pacientes y se ha estimado que perdura hasta al menos 17 días desde el inicio del tratamiento. Adicionalmente, en un pequeño ensayo de fase 3, avatrombopag probó su eficacia en adultos pre-tratados con TIPC grave, demostrando que el tratamiento prolonga en casi 12 semanas el tiempo que los pacientes mantienen un recuento de plaquetas de > 50.000/µl sin tratamiento de rescate (12,0 vs. 0,1 semanas con placebo).
Con respecto a la seguridad, son dos fármacos bien tolerados, con un perfil toxicológico más benigno que eltrombopag (el otro agonista del receptor de TPO no peptídico disponible), que se relaciona con un aumento del riesgo trombótico no confirmado para estos fármacos. Se ha notificado una frecuencia baja (< 10%) de reacciones adversas inespecíficas, con incidencia similar respecto a placebo. Para avatrombopag se han descrito como las más frecuentes: cefalea, náuseas, cansancio, mareos, dolor óseo, diarrea y pirexia; para lusustrombopag: cefalea, náuseas, trombosis de la vena porta y exantema. La amplia mayoría fueron leves y reversibles, no provocando discontinuaciones del tratamiento.
En definitiva, los dos nuevos fármacos no suponen una innovación reseñable en cuanto a mecanismo de acción, pero pueden superar los inconvenientes clínicos de la transfusión plaquetaria (tratamiento de elección), siendo las primeras opciones farmacológicas en pacientes hapatópatas con trombocitopenia grave que vayan a ser sometidos a un procedimiento invasivo programado a corto plazo. En ausencia de comparaciones directas entre ambos, no hay datos que permitan considerar la superioridad de uno sobre otro, posicionándose como alternativas similares en primera línea, si bien sus IPTs limitan su elección preferente a cuando no pueda hacerse la transfusión o en pacientes con numerosos procedimientos seguidos. En el tratamiento de TIPC, avatrombopag se posiciona como una opción alternativa a romiplostin o eltrombopag en 2ª línea, en pacientes que no responden al tratamiento preferente con corticosteroides o inmunoglobulinas (e incluso esplenectomía), de forma que parece incorporar un menor grado de innovación terapéutica en esta indicación.
Trombocitopenia inmunitaria crónica
Fostamatinib ▼TAVLESSE® (Instituto Griphols) PAM 447
Fostamatinib es un profármaco que ejerce su actividad a través de su metabolito principal en el que se convierte con rapidez a través de la fosfatasa alcalina intestinal: R406 actúa como un inhibidor potente y selectivo de la tirosina cinasa esplénica (SYK) e inhibe la transducción de señales de los receptores de los linfocitos B y de los receptores activadores de Fc, importantes en el inicio y propagación de respuestas autoinmunes celulares inducidas por anticuerpos. Así, el fármaco reduce el aclaramiento –vía fagocitosis– de las plaquetas circulantes mediado por los macrófagos en bazo e hígado. El medicamento ha sido autorizado para el tratamiento por vía oral de la trombocitopenia inmunitaria crónica (TIPC) en pacientes adultos que son resistentes a otros tratamientos.
Su autorización se ha sustentado en los resultados clínicos de dos ensayos pivotales (FIT-1 y FIT-2), aleatorizados, doble ciegos y controlados por placebo (N= 150), que incluyeron una amplia mayoría de pacientes con TIPC crónica, y con respuesta insuficiente a al menos una línea de tratamiento previo con corticoides, inmunoglobulinas, esplenectomía (un tercio) y/o un agonista del receptor de trombopoyetina (TPO). En el conjunto de la población, un tratamiento diario durante 24 semanas aportó una tasa de respuesta plaquetaria estable (recuento de ≥ 50.000/µl) significativamente más alta que con placebo: 16,8% (17/101) con fostamatinib frente al 2,1% (1/49) en el grupo placebo. Las variables secundarias relativas a los niveles sanguíneos de plaquetas a las semanas 12 y 24, comparativamente con el momento basal, respaldaron los objetivos primarios de eficacia. Además, la eficacia del fármaco fue consistente y de similar magnitud con independencia del número de líneas de tratamiento previo que hubieran recibido los pacientes (entre quienes habían fracasado a ≥ 3 terapias, se observó una respuesta plaquetaria estable en el 14% de los pacientes tratados con fostamatinib frente al 0% con placebo) y tras el cambio de tratamiento en pacientes con placebo inicialmente (en el estudio de extensión FIT-3). La respuesta plaquetaria tuvo una duración de > 1 año en dos tercios de los pacientes respondedores.
El perfil toxicológico para su uso en TIPC es importante, pero se considera clínicamente manejable con ajustes posológicos o farmacoterapia. Las reacciones adversas más frecuentemente notificadas en relación con el uso del fármaco fueron trastornos gastrointestinales (sobre todo, diarrea y náuseas), hipertensión, mareo y aumento de niveles de enzimas hepáticas; pero la mayoría fueron leves o moderadas en severidad y se resolvieron espontáneamente o con tratamiento. Las reacciones adversas graves fueron muy poco frecuentes (≤ 1%), y el uso del fármaco no se ha asociado con ninguna muerte ni con un riesgo específico de hemorragias.
Fostamatinib aporta cierto grado de innovación terapéutica: aunque su mecanismo de acción –inhibición de tirosina cinasa– es ampliamente conocido para muchos principios activos, inaugura una vía terapéutica en la indicación, con una novedosa diana terapéutica (la tirosina cinasa esplénica). Ha mostrado superioridad significativa frente a placebo en términos de respuesta plaquetaria, de especial interés en pacientes con TIPC refractaria que han agotado varias opciones terapéuticas, incluyendo esplenectomía, rituximab o agentes trombopoyéticos. Si bien en estos pacientes un efecto de magnitud incluso modesta se puede considerar importante, se aprecian importantes limitaciones para concluir sobre la relevancia clínica de los hallazgos. En este sentido, la ausencia de comparación con otros fármacos dificulta en gran medida el posicionamiento de fostamatinib: parece que puede tener un papel en el tratamiento de pacientes adultos a partir de una segunda línea de tratamiento inefectiva. Se debe esperar a tener seguimientos más prolongados de su uso para caracterizar el perfil beneficio-riesgo a largo plazo, como ya se conoce para los agonistas de receptores de TPO.
Angioedema hereditario
Conestat Alfa ▼RUCONEST® (Pharming Group NV) PAM 443
Lanadelumab ▼TAKHZYRO® (Shire Pharm.) PAM 443
Se trata de dos nuevos fármacos que actúan sobre la vía de señalización de la cascada de la coagulación y del complemento. Conestat alfa es un análogo recombinante del inhibidor de la C1-esterasa (INH-C1) humano obtenido en leche de conejas transgénicas que repone la actividad inhibitoria de esta proteína plasmática sobre distintas proteasas implicadas en los citados sistemas fisiológicos, que se encontrarían sobreactivados. Ha sido autorizado para el tratamiento por vía intravenosa de las crisis agudas de angioedema en adultos, adolescentes y niños (a partir de 2 años) con angioedema hereditario (AEH) debido a un déficit de INH-C1. Por otro lado, lanadelumab es un anticuerpo monoclonal IgG1 humano que se une específicamente e inhibe la actividad proteolítica de la calicreína plasmática, limitando la proteólisis de cininógeno y la liberación de bradicinina: evita el efecto de ésta en la promoción de la inflamación y la extravasación. El medicamento, designado como huérfano, ha sido aprobado para su administración por vía subcutánea en la prevención rutinaria de las crisis recurrentes de AEH en pacientes de ≥ 12 años de edad.
En dos estudios pivotales aleatorizados y doble ciego, conestat alfa ha demostrado una superioridad sobre placebo en el tratamiento agudo de las crisis de AEH, evidenciada en la reducción significativa del tiempo hasta el inicio del alivio de los síntomas de aproximadamente 120 min, y en el tiempo hasta la aparición de síntomas mínimos en comparación con placebo de unos 850 min. Si bien se excluyeron paciente con ataques graves y riesgo vital, el fármaco parece eficaz para abordar crisis en diferentes localizaciones anatómicas. Además, su perfil de seguridad/tolerabilidad es aceptable, sin diferencias en la frecuencia de reacciones adversas frente a placebo (las mayoritarias fueron cefalea, náuseas, molestias abdominales, vértigo, parestesias, e hinchazón en el punto de inyección) y con una escasa inmunogenicidad. En ausencia de comparaciones directas o indirectas de eficacia, el fármaco no supone ninguna innovación mecanística ni clínica respecto al inhibidor de C1-esterasa derivado de plasma actualmente disponible, con el que comparte perfil de seguridad (excepto la reacción a las proteínas de conejo) y ruta de administración. Tampoco parece aportar ninguna ventaja respecto al otro fármaco autorizado (icatibant, de administración subcutánea) y se posiciona como una alternativa de tratamiento similar a ellos.
Por otra parte, lanadelumab se ha autorizado en base a los resultados de otro estudio pivotal aleatorizado y doble ciego, en que un tratamiento de 26 semanas demostró ser significativamente más eficaz que placebo al reducir la tasa de ataques mensuales en pacientes con AEH en hasta un 87% (0,26-0,53 vs. 1,97). También redujo el número de crisis que precisaron tratamiento agudo, aumentando notablemente la proporción de pacientes sin crisis durante el tratamiento (31-44% vs. 2% con placebo). El beneficio se constató en todos los subgrupos de pacientes, se correlacionó con una mejoría clínicamente relevante en la calidad de vida reportada por los mismos, y parece mantenerse durante al menos un año de tratamiento. Aunque persisten incertidumbres de su seguridad e inmunogenicidad a largo plazo, el perfil toxicológico del fármaco es por lo general tolerable, y similar al del inhibidor de C1-esterasa; el principal evento adverso notificado fueron las reacciones en el sitio de inyección (52%) –dolor, eritema y hematoma–, leves y autolimitadas. Sin disponer de comparaciones directas o indirectas con el resto de opciones, lanadelumab se posiciona como una alternativa terapéutica al inhibidor de C1-esterasa en la profilaxis a largo plazo de las crisis de AEH, pero se asocia con un grado reseñable de innovación por incorporar un mecanismo de acción novedoso en la indicación y aportar la ventaja potencial de una mejora de la adherencia terapéutica por su pauta quincenal/mensual y su vía de administración subcutánea.
Antiinfecciosos de uso sistémico
Infecciones bacterianas complicadas
Meropenem / Vaborbactam ▼VABOREM® (Menarini) PAM 448
El nuevo medicamento consiste en la asociación de meropenem, un antibiótico betalactámico previamente comercializado del grupo de los derivados del carbapenem (cuya acción bactericida se media por la inhibición de la síntesis de la pared celular de peptidoglicano), y vaborbactam, un nuevo principio activo inhibidor de betalactamasas que amplía el espectro y la potencia antibacteriana del primero. Vaborbactam carece de núcleo betalactámico e inhibe las betalactamasas de clases A y C –incluida la carbapenemasa de K. pneumoniae– mediante la formación de un aducto covalente con las mismas, diferenciándose en estructura y selectividad de los otros inhibidores de betalactamasas disponibles (ácido clavulánico, tazobactam y avibactam); no protege a meropenem de las betalactamasas de clase B y D ni restaura la sensibilidad al carbapenémico si la resistencia se debe a la impermeabilidad de la membrana externa o a una bomba de eflujo. El medicamento ha sido autorizado para su uso por vía intravenosa, siguiendo las directrices oficiales del uso apropiado de antibacterianos, para el tratamiento en pacientes adultos de infecciones urinarias complicadas (incluida la pielonefritis), infecciones intraabdominales complicadas, neumonías intrahospitalarias (incluida la asociada a ventilación mecánica), bacteriemias asociadas –o con sospecha de estar asociadas– a cualquiera de las infecciones anteriores e infecciones debidas a microorganismos gran negativos aerobios en adultos con opciones de tratamiento limitadas. Se ha probado su eficacia clínica frente a patógenos como Escherichia coli, Klebsiella pneumoniae y el complejo Enterobacter cloacae spp, pero no se ha confirmado aún su actividad frente a otras bacterias potencialmente implicadas en las indicaciones aprobadas.
Los datos clínicos procedentes de dos estudios clínicos de fase 3 han contrastado adecuadamente la eficacia de la combinación a las dosis autorizadas en su indicación en ITUc y pielonefritis. Los resultados revelaron su no inferioridad frente a piperacilina/tazobactam: en la visita de curación entre los días 15 y 23 de tratamiento, la tasa de erradicación microbiológica era del 67% en el brazo experimental frente al 58% en el brazo control (el beneficio fue mayor en pacientes con pielonefritis aguda que en ITU complicadas con fuente de infección removible). Pero es importante subrayar que por ahora no se dispone de datos definitivos de eficacia en el resto de las indicaciones (uno de los estudios solo incluyó un número muy limitado de pacientes con dichas infecciones), sino que la autorización se basa en la experiencia con la monoterapia con meropenem, datos de preclínica y análisis combinados de farmacocinética/farmacodinamia, aceptando que se pueden alcanzar concentraciones de vaborbactam en la cavidad abdominal y en el epitelio pulmonar suficientes para proteger a meropenem frente a carbapenemasas y betalactamasas. En términos de seguridad, el perfil toxicológico del medicamento se puede considerar similar al de los comparadores activos, esto es, un perfil relativamente benigno, manejable, y equiparable al de otros antibióticos betalactámicos. Las reacciones adversas más comunes durante el tratamiento son la cefalea (8%), la diarrea (5%), flebitis en el lugar de perfusión (2%) y las náuseas (2%). Ninguna de ellas fue considerada grave o motivo de interrupción del tratamiento (la tasa de discontinuación fue baja, en torno al 1%).
En resumen, se trata la primera combinación de un antibiótico carbapenémico con un inhibidor de betalactamasas, que puede jugar un papel importante en el abordaje de un grave problema de salud pública como son las infecciones causadas por enterobacterias (entre ellas, K. peumoniae) y Pseudomonas aeruginosa multirresistentes productoras de betalactamasas de espectro extendido o de carbapenemasas. Es decir, se trata de un buen antibiótico que amplía las opciones disponibles para varios tipos de infecciones complicadas, pero no aporta ningún elemento innovador farmacológico ni clínico respecto a otras opciones ya disponibles. El IPT lo posiciona como una alternativa en la terapéutica dirigida cuando se disponga del antibiograma, de utilidad también en el tratamiento empírico en casos de sospecha o alta prevalencia local de enterobacterias productoras de carbapenemasas A; pero no debe ser una opción preferente en el tratamiento empírico de infecciones por enterobacterias productoras de betalactamasas distintas, pues existen otras alternativas. Su elección se hará de forma individualizada, preferiblemente dentro de programas de optimización de uso de antibióticos.
Infección por SARS-CoV-2: COVID-19
Remdesivir ▼VEKLURY® (Gilead) PAM 442
Remdesivir es un nuevo agente antiviral profármaco del nucleótido de adenosina, que se metaboliza en el interior de las células humanas dando lugar a un metabolito trifosfato farmacológicamente activo análogo de nucleósido, el cual inhibe selectivamente la ARN polimerasa dependiente de ARN del SARS-CoV-2 por competir por similitud con el sustrato natural (ATP) en la incorporación en las cadenas de ARN nacientes. Como consecuencia, se produce la terminación retardada de la cadena del ARN en formación durante la replicación viral. El medicamento ha sido aprobado para el tratamiento por vía intravenosa de la COVID-19 en adultos y adolescentes (de 12 años y mayores con un peso corporal de al menos 40 kg) con neumonía que requieren oxígeno suplementario.
Ha alcanzado la autorización condicional en la UE gracias a los datos derivados del ensayo pivotal aleatorizado ACCT-1, en el cual los pacientes hospitalizados con COVID-19 tratados con remdesivir a la dosis autorizada durante un máximo de 10 días (N= 541) experimentaron una mejoría clínica casi un tercio más rápida que aquellos que recibieron placebo (N= 522), acortando la mediana del periodo hasta la recuperación en 5 días (10 vs. 15 días; RR= 1,29; p< 0,001). Esa eficacia fue más marcada en el subgrupo mayoritario de pacientes con enfermedad grave, en quienes la mediana del tiempo hasta la recuperación se redujo 6 días (12 vs. 18 días con placebo; RR= 1,37; p< 0,001). En cambio, no hubo un beneficio significativo en pacientes con enfermedad leve-moderada o en aquellos que estaban más críticos al inicio del estudio (necesidad de ventilación mecánica invasiva u oxigenación por membrana extracorpórea). Además, los datos del citado estudio y del estudio RECOVERY impulsado por la OMS sugieren que, aunque la tendencia pueda ser favorable, no se ha probado un beneficio en mortalidad a 28 días con el uso del fármaco. Con respecto a la seguridad, el perfil toxicológico de remdesivir es relativamente benigno, con una tolerabilidad similar a placebo y una tasa de discontinuación por eventos adversos baja (< 10%). Así, los eventos adversos graves son menos frecuentes que con placebo (25% vs. 32%), destacando un menor número de casos de fallo respiratorio severo (5% vs. 8%). También sobresalen por su frecuencia: anemia, insuficiencia renal aguda, pirexia, hiperglucemia y elevación de los niveles de transaminasas hepáticas, siendo esta última la reacción adversa posiblemente relacionada con el tratamiento más frecuente en voluntarios sanos, mientras que en pacientes con COVID-19 tratados predominan las náuseas. Se debe tener precaución con el uso del fármaco en pacientes con cualquier grado de insuficiencia hepática o renal.
Todo apunta a que remdesivir, sin representar una cura radical, tiene una eficacia global modesta como antiviral en la COVID-19 grave, aportando el mayor beneficio en tratamientos de una duración máxima de 5 días en un subgrupo específico de paciente que requieren suplementación con O2 exógeno. Constituye una innovación disruptiva de la investigación farmacológica por tratarse del primer fármaco en ser autorizado frente a la COVID-19 en la UE, aprobado tras poco más de 6 meses desde la identificación del agente causal de una patología infecciosa que representaba una necesidad médica no cubierta y que se ha asociado a una carga sanitaria y socio-económica sin precedentes en el mundo moderno; en ese contexto de sobrecarga del sistema sanitario, se prevé que tenga un impacto positivo en términos de alivio de síntomas y ahorro y optimización de recursos. Los resultados clínicos fueron evaluados por la EMA mediante un proceso de evaluación continua excepcionalmente rápido, que sirvió de base para la aceleración de los procesos regulatorios (rolling reviews) que ha predominado posteriormente, por ejemplo, en la autorización de vacunas. Aunque los beneficios superan a los riesgos derivados de las incertidumbres existentes, aún permanecen dudas sobre el beneficio real de remdesivir: se desconoce si afecta a la carga viral y la capacidad de contagio, y se duda de si podría haber un mayor beneficio por su empleo en fases más precoces de la infección.
Vacuna de ARNm
▼COMIRNATY® (Pfizer/BioNTech) PAM 440
▼SPIKEVAX® (Moderna) PAM 440
BNT162b2 (Comirnaty®) y mRNA-1273 (Spikevax®) son dos nuevas vacunas frente a la COVID-19 en las que el principio activo es un ARNm monocatenario, codificante para una variante de la proteína S del SARS-CoV-2 de cadena completa y estabilizada en conformación prefusión, que se formula en nanopartículas lipídicas que lo protegen y facilitan su entrada en las células hospedadoras humanas. La traducción transitoria de la proteína S en el ribosoma permitirá su expresión en la membrana de células presentadoras de antígenos, donde será reconocida como antígeno extraño por las células del sistema inmunitario, desencadenando una respuesta específica de anticuerpos neutralizantes y de inmunidad celular, que contribuirá a la protección frente a COVID‑19 sintomática ante una futura exposición al SARS-CoV-2. En base a ello, ambos medicamentos han sido autorizados en una pauta de 2 dosis intramusculares para la inmunización activa para prevenir la COVID‑19 causada por el SARS‑CoV‑2, con la diferencia de la edad: BNT162b2 en personas de ≥ 16 años de edad y mRNA-1273 en personas de ≥ 18 años. Ambos medicamentos deben utilizarse conforme a las recomendaciones oficiales.
La autorización de BNT162b2 ha derivado de un amplio ensayo pivotal, multicéntrico, controlado por placebo y simple ciego, de dos brazos de tratamiento, que ha enrolado a más de 43.500 participantes de ≥ 12 años. Con una mediana de seguimiento de 2 meses en más de 36.600 participantes, demostró una tasa de eficacia global estimada en el 95,0% (p< 0,001): solo se registraron 8 nuevos casos confirmados de COVID-19 tras ≥ 7 desde la 2ª dosis en sujetos sin evidencia de infección previa por SARS-CoV-2 tratados con la vacuna (N= 18.195; 0,04%), frente a los 162 casos notificados en el grupo placebo (N= 18.325; 0,88%). Por su parte, el estudio pivotal que condujo a la autorización de mRNA-1273 aleatorizó en dos brazos de tratamiento a más de 30.400 participantes adultos (≥ 18 años) y, con una mediana de seguimiento de 92 días, se estimó una eficacia global de la vacuna fue del 94,1% (p< 0,001): se confirmó COVID-19 sintomática de inicio ≥ 14 días desde de la 2ª dosis en solo 11 participantes tratados con la vacuna (N= 14.134; 0,08%) frente a los 185 casos registrados en el grupo placebo (N= 14.073; 1,31%). Para ambas vacunas, la eficacia se reveló consistente e independiente de factores como la edad, sexo, raza, comorbilidades o antecedentes de infección por SARS-CoV-2, aunque aún se plantean incertidumbres relativas a su eficacia para prevenir la COVID-19 grave, frente a la infección asintomática o sobre la duración de la inmunidad. Respecto a su seguridad, son dos vacunas bien toleradas a corto plazo, con un perfil toxicológico definido fundamentalmente por reacciones adversas locales leves-moderadas y transitorias (desaparecen en 1-3 días), destacando el dolor y la hinchazón en el punto de inyección, menos frecuentes a mayor edad de la persona. Entre las reacciones adversas sistémicas sobresalen la fatiga, la cefalea y las mialgias/artralgias, si bien la frecuencia de eventos adversos graves es baja y similar entre vacunas y placebo. La caracterización de la seguridad a medio y largo plazo aún requiere esperar a los resultados del seguimiento durante 24 meses, previsto en los ensayos clínicos.
Además de ser las primeras opciones disponibles de profilaxis farmacológica frente a COVID-19, incorporan una tecnología completamente innovadora en vacunas de uso humano, abriendo la puerta a la probable llegada de futuras vacunas a base de ARNm. Han demostrado elevada eficacia para prevenir la COVID-19 sintomática (94-95%) y buena tolerabilidad a corto plazo. Por el impacto potencial que pueden tener en la salud pública a nivel global en una pandemia de elevado impacto sanitario y socioeconómico, representan indudablemente una innovación terapéutica disruptiva. El propio proceso excepcionalmente rápido hasta la autorización condicional de comercialización de estas vacunas constituye un hito por sí mismo, cumpliendo las garantías adecuadas de calidad, seguridad y eficacia.
Vacuna recombinante ChAdOx1-S
▼VAXZEVRIA® (AstraZeneca) PAM 441
La vacuna monovalente ChAdOx1-S está compuesta por un vector viral de adenovirus de chimpancé, recombinante y no replicativo, que contiene una secuencia de ADN codificante para la glicoproteína S del SARS-CoV-2, en su conformación trimérica prefusión (no modificada para una mayor estabilización). La expresión transitoria de la proteína S por las células huésped en la zona de inyección permitirá que sea reconocida como antígeno extraño por las células del sistema inmunitario, desencadenando una respuesta adaptativa tanto de anticuerpos neutralizantes como de inmunidad celular por linfocitos T, que contribuirán a la protección frente a COVID‑19 sintomática ante una futura exposición al SARS-CoV-2. En base a ello, el medicamento ha sido aprobado para la inmunización activa para prevenir la COVID-19 causada por SARS-CoV-2, en personas de 18 años de edad y mayores en una pauta de dos dosis intramusculares separadas entre 4 y 12 semanas (28 a 84 días), debiéndose usar siempre siguiendo las recomendaciones oficiales.
La autorización condicional de ChAdOx1-S ha derivado de los resultados de un análisis preliminar conjunto de dos de los estudios de fase 3 (N= 11.636; 6.106 participantes recibieron 2 dosis separadas entre 4 y 12 semanas). Tras la confirmación de 64 casos de COVID-19 sintomática en el grupo de la vacuna y 154 casos en el grupo control, se determinó una eficacia protectora del 59,5% en el global de participantes (del 62,6% entre quienes recibieron las dos dosis completas, y de hasta el 90,0% en los participantes que recibieron una primera media dosis). Un análisis posterior con más datos (N> 17.000) ha confirmado una eficacia global del 66,7% en la prevención de casos de COVID-19 sintomática tras ≥ 15 días desde la segunda inyección, independientemente de que la primera dosis sea media o completa. La eficacia de la vacuna se mostró independiente de factores como género, raza o comorbilidades y fue del 100% en la prevención de casos graves de COVID-19 a partir de las 3 semanas desde la primera dosis. Sin emabargo, no se pudo concluir sobre la eficacia en función de la edad, ya que fueron muy pocos los sujetos de > 55 años reclutados (< 5% de personas con ≥ 70 años) y, en consecuencia, el número de casos confirmados en ellos. También persisten incertidumbres respecto al tiempo de inmunidad conferida, la capacidad de prevenir el ingreso en UCI o la mortalidad, la eficacia en infección asintomática o la capacidad de contagio, o frente a las nuevas variantes virales. Con respecto a la seguridad, se trata de una vacuna muy bien tolerada a corto plazo: el perfil toxicológico se define por reacciones adversas locales de gravedad leve-moderada y transitorias, destacando la sensibilidad y el dolor en el punto de inyección, más leves y menos frecuentes a mayor edad de la persona. Entre las reacciones adversas sistémicas sobresalen por su frecuencia: cefalea, fatiga, mialgia, malestar, pirexia y escalofríos, o artralgia. En cualquier caso, la frecuencia de eventos adversos graves es baja y muy similar a placebo. Habrá que esperar a los datos de seguridad tras el seguimiento de 1 año previsto en los estudios a fin de caracterizar la seguridad a medio y largo plazo, así como esclarecer los riesgos de su uso en embarazo.
Por tratarse de la primera vacuna que se autoriza no basada en ARNm representa cierto grado de innovación farmacológica en la indicación, si bien el uso de vectores virales no es una tecnología o estrategia vacunal tan pionera como lo ha sido el ARNm. Con un perfil toxicológico y pauta posológica similares, parece que induce una inmunoprotección notablemente menos eficaz que las vacunas de ARNm BNT162b2 (Comirnaty®) y mRNA-1273 (COVID-19 Vaccine Moderna®). Ello, unido a la incertidumbre sobre su uso en adultos mayores, sugiere que esta vacuna ChAdOx1-S no supone un avance farmacoterapéutico disruptivo, pero contribuye a ampliar el arsenal terapéutico para elevar las coberturas vacunales en un contexto de emergencia sanitaria mundial con una principal ventaja logística respecto a las otras dos vacunas: no requiere congelación y se puede conservar hasta 6 meses en nevera (2-8ºC).
Vacuna recombinante Ad26.COV2-S
▼COVID-19 Vaccine Janssen® (Janssen) PAM 443
Esta vacuna está compuesta por un vector de adenovirus humano tipo 26, recombinante y no replicativo, que contiene una secuencia de ADN codificante para la glicoproteína S del SARS-CoV-2 de longitud completa, en una conformación prefusión estabilizada mediante mutaciones específicas. La expresión transitoria de la proteína S por las células huésped en la zona de inyección permitirá que sea reconocida como antígeno extraño por las células del sistema inmunitario, desencadenando una respuesta adaptativa tanto de anticuerpos neutralizantes como de inmunidad celular por linfocitos T, que contribuirán a la protección frente a COVID-19 sintomática ante una futura exposición al SARS-CoV-2. En base a ello, el medicamento ha sido aprobado para la inmunización activa para prevenir la COVID-19 en personas de 18 años de edad y mayores en una pauta dosis única por vía intramuscular, debiéndose usar siempre siguiendo las recomendaciones oficiales.
Su autorización condicional se sustentó en los resultados del análisis de eficacia de un amplio estudio pivotal controlado de fase 3 (N > 44.000 adultos, casi 21.900 recibieron la vacuna) realizado en EE.UU., Latinoamérica y Sudáfrica. Con un seguimiento de unos dos meses, tras la confirmación de 116 casos de COVID-19 sintomática en el grupo de la vacuna y 348 casos en el grupo placebo, se determinó una eficacia protectora global en sujetos seronegativos para el SARS-CoV-2 del 66,9% tras 14 días desde la vacunación. Esa protección se mantuvo en el 66,1% en los casos de inicio después de ≥ 28 días. La eficacia se mostró independiente de factores como el sexo, la presencia de comorbilidades, e incluso de la edad, aportando una inmunoprotección superior en sujetos de mayor edad: tras ≥ 28 días, la eficacia vacunal fue del 65% entre 18 y 64 años y de hasta el 74% en participantes de ≥ 65 años. Además, un análisis exploratorio por regiones geográficas sugirió que su eficacia, aunque ligeramente menor, se mantiene en niveles elevados frente a las variantes virales B.1.351 y P.2, con una protección del 64% en Sudáfrica, el 68% en Brasil y el 72% en EE.UU. De modo interesante, la eficacia frente a la enfermedad grave fue de > 85% tras 28 días desde la vacunación, no registrándose ninguna muerte por COVID-19 entre las personas vacunadas; ese efecto fue consistente entre regiones geográficas. Persisten incertidumbres en torno a la eficacia de la vacuna en mayores de 75 años, la prevención de ingreso en UCI o mortalidad, frente a la infección asintomática y la capacidad de transmitir la enfermedad o sobre el tiempo de inmunidad conferida. Con respecto a la seguridad, se trata de una vacuna bien tolerada a corto plazo: el perfil toxicológico se define por reacciones adversas de gravedad leve-moderada y transitorias, destacando el dolor en el punto de inyección, la cefalea, la fatiga, mialgias y náuseas. Habrá que esperar a los datos tras el seguimiento de 2 año previsto en el ensayo pivotal para caracterizar su seguridad a medio-largo plazo, no pudiéndose descartar aún la posibilidad de anafilaxia ni de aparición de eventos trombóticos raros.
En el actual contexto de pandemia, con la aparición de nuevas variantes virales y la necesidad de disponer de suficientes medicamentos profilácticos autorizados, esta vacuna es la primera que se autoriza en pauta de 1 dosis, representando un cierto grado de avance terapéutico en su indicación, si bien no supone una innovación farmacológica disruptiva (el uso de vectores adenovirales no es una estrategia tan novedosa como lo fue el ARNm). Con un perfil de seguridad similar al resto de vacunas aprobadas en la UE, parece que aporta una inmunoprotección inferior a la de las vacunas de ARNm, que se mueven en tasas de eficacia del 94-95%, pero similar o superior a ChAdOx1-S (Vaxzevria®), cercana al 60%. Además, el grado de prevención de COVID-19 grave (> 85%) es muy relevante clínicamente. Contribuirá a ampliar el arsenal terapéutico para elevar las coberturas vacunales, aportando como ventaja –respecto a las de ARNm– su mayor estabilidad: no requiere congelación y se puede conservar 3 meses en nevera (2-8ºC) e incluso 12 horas a temperatura ambiente.
Infección por citomegalovirus
Letermovir ▼PREVYMIS (MSD) PAM 449
Es un nuevo antiviral que se dirige específicamente frente a citomegalovirus (CMV). Actúa a través de la inhibición del complejo ADN terminasa viral: por su mecanismo –interferencia con la formación de unidades monoméricas de genoma de longitud adecuada y con la maduración y ensamblado de los viriones– se diferencia del resto de antivirales anti-CMV disponibles (ganciclovir, valganciclovir, cidofovir y foscarnet), que son inhibidores de la ADN polimerasa, con una posible mejor barrera genética y sin esperarse resistencias cruzadas. El medicamento, designado como huérfano, ha sido autorizado para la profilaxis por vía oral e intravenosa de la reactivación del CMV y de la enfermedad causada por este virus en adultos seropositivos para el CMV [R+] receptores de un trasplante alogénico de células madre hematopoyéticas (TPH).
Dicha indicación se ha sustentado en los datos de un ensayo pivotal de fase 3 (N= 565), bien diseñado, en que una profilaxis diaria –iniciada una mediana de 9 días desde el TPH– ha demostrado que letermovir es significativamente superior a placebo: reduce en 23,5 puntos porcentuales la tasa de fracaso a la semana 24 (37,5% vs. 60,6%; p< 0,0001), disminuyendo un 60% la necesidad de inicio de terapia anticipada por infección clínicamente significativa por CMV (18% vs. 42%); en cambio, la tasa de desarrollo de enfermedad orgánica por CMV y necesidad de tratamiento específico fue baja y similar en ambos grupos (1,5% vs. 1,8%). Los resultados de las variables secundarias (reducción de 31,3 puntos porcentuales en la tasa de fracaso a la profilaxis en la semana 14) respaldaron la eficacia, que se mostró consistente en todos los subgrupos de pacientes. Sin embargo, persisten incertidumbres respecto a la durabilidad del efecto del tratamiento y sobre su efecto en la supervivencia a largo plazo. Por otra parte, el perfil toxicológico del fármaco, bien definido, parece aceptable, sin diferencias relevantes respecto a placebo en la frecuencia global de eventos adversos ni en la proporción de pacientes con efectos adversos graves. Con su uso predominan las reacciones adversas relacionadas con el tracto gastrointestinal, tales como diarrea, náuseas y vómitos, y otros menos frecuentes (trastornos cardiacos, disnea, mialgias o desórdenes laberinto/oído); en su mayoría son leves-moderadas, de modo que la tasa de interrupción del tratamiento por problemas de seguridad es baja (4,8% vs. 3,6% con placebo). Además, es preciso destacar que el fármaco no induce mielosupresión, de relevancia en pacientes sometidos a un TPH.
En definitiva, se trata del primer fármaco autorizado en la UE para la profilaxis de la reactivación de CMV en adultos seropositivos para dicho virus que reciben un TPH alogénico. Se ha probado que reduce la tasa de fracaso de la profilaxis frente a placebo y a más de la mitad la necesidad de iniciar terapia anticipada con otros antivirales (ganciclovir/valganciclovir), pero sin diferencias en el desarrollo de la enfermedad orgánica por CMV, aspecto que podría considerarse clínicamente más relevante; tampoco se pueden extrapolar los resultados a subgrupos de pacientes no investigados (infección por VIH, hepatitis, uso de alemtuzumab, etc). Puede ser útil para evitar los efectos mielosupresores de la terapia anticipada en la primera fase del trasplante, con especial beneficio en pacientes con mayor riesgo de reactivación de CMV, pero las incertidumbres en los datos de eficacia limitan sustancialmente el grado de innovación terapéutica, a pesar de incorporar un novedoso mecanismo de acción. También se debe tener en cuenta que es un fármaco específico anti-CMV, sin actividad contra otros virus frecuentes en receptores de TPH, por lo que habría que continuar usando otros antivirales si así se requiere.
—En 2021 sobresalió la comercialización de los dos primeros medicamentos de ARNm para prevenir la COVID-19 (Comirnaty®
y Spikevax®)—
Agentes antineoplásicos e inmunomoduladores
Leucemias
Linfocitos T transducidos con CAR ANTI-CD19 (A3B1) 4-1BB/CD3ζ) ▼ARI-0001 (Hospital Clinic de Barcelona) PAM 444
ARI-0001 es un novedoso medicamento de terapia avanzada cuyo principio activo son linfocitos T autólogos transducidos ex vivo para expresar un receptor de antígeno quimérico (CAR) con especificidad anti-CD19. Tras la unión de los linfocitos T reprogramados a las células que expresan CD19 –células del linaje B, tanto malignas como normales–, la proteína quimérica transmite las señales intracelulares necesarias para ejercer la actividad antineoplásica, así como una señal que favorece su expansión y persistencia. La AEMPS ha otorgado al Hospital Clinic de Barcelona la autorización de uso para su fabricación y administración –en perfusión intravenosa fraccionada– en el tratamiento de leucemia linfoblástica aguda (LLA) de células B CD19+ en recaída o refractariedad tras ≥ 2 líneas de tratamiento o en recaída postrasplante en pacientes adultos de > de 25 años. Es la 2ª autorización con cláusula de exención hospitalaria (la primera para una terapia con células modificadas genéticamente) tras el medicamento NC1 del Hospital Universitario Puerta de Hierro de Majadahonda, autorizado en 2019 para el tratamiento de lesiones medulares traumáticas.
En un estudio piloto de fase 1, los pacientes adultos de > 25 años con LLA-B en recaída o refractariedad que recibieron la infusión tras su inclusión (n= 9), mostraron una tasa de respuesta global (TRG) del 67%: 56% de respuesta completa (RC) y 11% de respuesta completa con recuperación hematológica incompleta (RCi). La mediana de la duración de la respuesta (DR) entre los respondedores fue de > 6 meses, con datos sugerentes de una respuesta más prolongada en algunos de ellos. Considerando todos los pacientes infundidos de > 25 años (n= 19), incluidos aquellos en RC al inicio, la TRG al día 100 fue del 79%, la mediana de la supervivencia sin progresión alcanzó los 7,2 meses y la DR los 8,7 meses; así, más de la mitad de los pacientes (51%) estaban vivos al año desde la infusión. Con respecto a la seguridad, el importante perfil toxicológico del medicamento es cualitativa y cuantitativamente similar al descrito para otras terapias CAR-T evaluadas en LLA: se notifican eventos adversos relacionados con el medicamento y posteriores a su perfusión en la práctica totalidad de pacientes, siendo graves (grado ≥ 3) o potencialmente mortales en un tercio de ellos. Destacan, por su frecuencia y gravedad, el síndrome de liberación de citoquinas (55%, 13% graves), las infecciones como consecuencia de las citopenias (55%, 5% de neumonía grave) y los eventos adversos neurológicos (442%, 3% graves) como los aspectos cruciales del manejo de su toxicidad. Otras reacciones adversas comunes son: elevación de enzimas hepáticas, vómitos, cefalea, pirexia y disminución del apetito. Persisten incertidumbres sobre el riesgo a largo plazo de desarrollo de neoplasias secundarias, reactivaciones virales, etc.
A fin de posicionar el medicamento, conviene recordar que las terapias hasta ahora empleadas en práctica clínica en LLA-B en refractariedad/recaída se asocian, en términos generales, con tasas de RC del 30-45% y medianas de supervivencia global de entre 4 y 8 meses. Todo parece indicar que los resultados con ARI-0001 son superiores a los comentados, y al menos comparables a los de terapias similares que, por su fundamento y beneficio clínico, supusieron una auténtica revolución terapéutica en población pediátrica con patología oncohematológica (tisagenlecleucel). ARI-0001 cubre la laguna terapéutica existente en adultos mayores de 25 años con LLA-B refractaria, de pronóstico muy pobre y para quienes no había ninguna opción específicamente autorizada, y profundiza en el prometedor campo de los medicamentos CAR-T. Entre sus aspectos innovadores sobresale el hecho de que se trata de la primera terapia de fabricación no industrial, con un desarrollo y producción plenamente europeos. Incorpora como ventaja potencial frente a otros CAR-T comerciales la rapidez con que puede desarrollarse el proceso –en un mismo hospital– desde la aféresis de células T hasta la administración del medicamento. Los limitados periodos de seguimiento y la escasa experiencia postautorización hasta la fecha impiden confirmar aún su potencial como tratamiento curativo, debiéndose esperar a los resultados de futuros estudios para verificar si existe beneficio en términos de supervivencia global a largo plazo.
Acalabrutinib ▼CALQUENCE (AstraZeneca) PAM 447
Acalabrutinib es un nuevo inhibidor potente y altamente selectivo de la tirosina cinasa de Bruton (BTK), una enzima que participa en la señalización bioquímica del receptor de antígenos y del receptor de citocinas de las células B, favoreciendo la supervivencia y proliferación de linfocitos B tumorales. Así, la inhibición covalente e irreversible que inducen acalabrutinib y su metabolito ACP-5862 sobre el centro activo de la enzima se traduce en una inhibición de la adhesión y migración dependiente de integrinas de linfocitos B, conducente a la reducción tumoral. El medicamento ha sido autorizado para el tratamiento por vía oral (100 mg/2 veces al día) en monoterapia o en combinación con obinutuzumab de pacientes adultos con leucemia linfocítica crónica (LLC) no tratados previamente, y para su uso en monoterapia en pacientes adultos con LLC que han recibido ≥ 1 tratamiento previo.
Su aprobación se sustentó en los datos clínicos de dos estudios pivotales aleatorizados y multicéntricos, de fase 3, abiertos, simple ciego y de grupos paralelos. En uno de ellos (ELEVATE-IN), con 535 pacientes adultos con LLC no tratados previamente, la asociación acalabrutinib+obinutuzumab y la monoterapia de acalabrutinib mostraron una superioridad notable frente a una combinación de obinutuzumab y clorambucilo (control activo). Tras 28 meses, sin alcanzarse la mediana de SLP en los brazos del fármaco (vs. 22,6 meses en el grupo control), la administración de acalabrutinib con o sin obinutuzumab redujo en un 90% y un 80% el riesgo de progresión o muerte por la enfermedad, respectivamente; se alcanzaron estimaciones de SLP a 24 meses del 93% y del 87%. El otro estudio (ASCEND) incluyó a pacientes adultos con LLC refractaria o en recaída tras ≥ 1 tratamiento previo (N= 310). Tras 16 meses, la monoterapia con acalabrutinib demostró una eficacia superior al régimen activo usado como control (idelalisib + rituximab o bendamustina + rituximab): tampoco se alcanzó la mediana de SLP en el brazo con el nuevo fármaco (vs. 16,5 meses), y se estimó una reducción del 69% en el riesgo de muerte o progresión. En general, la eficacia de acalabrutinib se reveló duradera y consistente en pacientes con factores citogenéticos de alto riesgo. Los datos de supervivencia global (aún inmaduros) y de tasa de respuesta reafirman la utilidad clínica del fármaco tanto en pacientes naïve como pretratados. En términos de seguridad, tiene un perfil toxicológico notable, con frecuentes y relevantes eventos adversos en los dos tipos de pacientes. La incidencia de eventos adversos de intensidad de grado 3-4 fue del 50-70% y supone tasas de discontinuación del tratamiento del 10-11% y de reducción de dosis del 4-7%. Las reacciones adversas más frecuentes (> 20%) fueron infección, dolor de cabeza, diarrea, hematomas, dolor musculoesquelético, náuseas, fatiga, tos y erupción cutánea. Entre las más graves, sobresalen las infecciones y las citopenias; pero tampoco se puede obviar el riesgo aumentado de segundas neoplasias primarias. No se observaron diferencias relevantes de seguridad en pacientes de ≥ 65 años o en aquellos con perfil citogenético de peor pronóstico.
Se trata de un inhibidor de BTK de 2ª generación cuya mayor selectividad por la enzima hace que sea al menos tan eficaz como ibrutinib pero con un perfil de toxicidad mejorado; presenta, por ejemplo, un menor riesgo de eventos adversos off target a nivel cardiaco (por ejemplo, de generación de arritmias). Sin embargo, no se dispone aún de comparaciones entre ambos, y habrá que esperar a un estudio ahora en marcha en pacientes con LLC en recaída/refractariedad y alto riesgo citogenético para confirmar sus diferencias en eficacia y seguridad. Por ahora, es probable que el uso de acalabrutinib, tanto en monoterapia como asociado a obinutuzumab, se considere una opción preferencial en primera línea para los pacientes con LLC no tratados, similar al uso de ibrutinib o venetoclax (mayoritariamente asociados a obinutuzumab). En pacientes con LLC pretratados la monoterapia con acalabrutinib viene a completar las posibilidades de tratamiento junto a ibrutinib o venetoclax más rituximab, aportando una respuesta clínica relevante en cuadros con escasas opciones terapéuticas, incluso cuando éstas no son funcionales. A la vista de lo demostrado para ibrutinib, es previsible que en un futuro se pruebe un beneficio clínico con acalabrutinib en otras neoplasias de células B.
Linfomas
Polatuzumab vedotina ▼POLIVY (Roche) PAM 450
Se recomienda consultar el artículo monográfico sobre polatuzumab vedotina que se publica en este mismo número.
Mogamulizumab ▼POTELIGEO (Kyowa Kyrin) PAM 449
Mogamulizumab es un nuevo anticuerpo monoclonal de tipo IgG1К que se une selectivamente al receptor CCR4 (del inglés C-C chemokine receptor type 4), el cual se expresa de forma inherente en la superficie de ciertas células cancerosas, entre las que se incluyen las de linfomas cutáneos de linfocitos T (LCCT), como la micosis fungoide y el síndrome de Sézary. CCR4 es un receptor acoplado a proteína G de ciertas quimiocinas CC que participan en la circulación de linfocitos a diversos órganos, incluida la piel. Al unirse al receptor, mogamulizumab impide la acción biológica de las quimiocinas y, por mecanismos de citotoxicidad celular dependiente de anticuerpos, provoca la depleción de las células diana. El medicamento, designado como huérfano, ha sido autorizado para el tratamiento –por vía intravenosa– de pacientes adultos con micosis fungoide (MF) o síndrome de Sézary (SS) que han recibido como mínimo un tratamiento sistémico previo.
Su aprobación se basó en un estudio pivotal abierto de fase 3, controlado por vorinostat, en el que un tratamiento con mogamulizumab en pacientes con MF y SS (N= 372) pretratados con alguna terapia sistémica (mayoritariamente bexaroteno, interferón y/o metotrexato) indujo una prolongación de 4,6 meses en la mediana de SLP (7,7 vs. 3,1 meses con vorinostat), reduciendo en un 47% el riesgo de progresión o muerte por la enfermedad; por subgrupos, el mayor beneficio se observó en aquellos con patología en estadios más avanzados (10,9 vs. 3 meses; HR= 0,36) y con SS (13,3 vs. 3,1 meses; HR= 0,32). Las tasas de respuesta fueron bajas comparativamente con lo conocido para otras alternativas usadas en práctica clínica, pero el nuevo fármaco indujo una TRG significativamente mayor que el control activo (28% vs. 4,8%) y una duración de la respuesta considerable (14,1 vs. 9,1 meses); la respuesta se observó en el compartimento sanguíneo (67% vs. 18%). No se ha confirmado hasta el momento un aumento en supervivencia global. Con respecto a la seguridad, se trata de un fármaco con un perfil de seguridad caracterizado fundamentalmente por eventos adversos de carácter leve-moderado y manejables clínicamente con ajustes posológicos. Las reacciones adversas al tratamiento más frecuentes fueron las relacionadas con la infusión (33%; 1,6% de grado ≥ 3), infecciones (24%; 9% de grado ≥ 3) y erupción medicamentosa (23%; 4,4% de grado ≥ 3). Algunas investigaciones apuntan a un mayor riesgo de complicaciones tras un trasplante de progenitores hematopoyéticos si se administra el nuevo fármaco previamente, quizás debido al agotamiento en sangre de las células Treg que provoca; por ello, aún se debe caracterizar mejor la seguridad en pacientes potencialmente candidatos a un TPH, y hasta entonces debería evitarse su uso en quienes se prevea.
No se dispone de comparaciones directas de mogamulizumab con otras alternativas usadas en 2ª línea y posteriores del tratamiento de pacientes con MF o SS (bexaroteno, interferón, metotrexato, gemcitabina, etc.). Las comparaciones indirectas, de robustez limitada, sugieren que brentuximab vedotina puede tener mejor perfil beneficio-riesgo en pacientes con LCCT CD30+. Se trata del primer fármaco para el que se aprueba la indicación específica de tratamiento de la MF y el SS y el primer agente biológico específicamente dirigido a CCR4 comercializado en Europa: inaugura una vía terapéutica con una potencial aplicabilidad en otros tipos de tumores, para los que ya se está investigando clínicamente. En un contexto en que los pacientes con MF y SS pueden sufrir síntomas agresivos y el estigma social de tener lesiones cutáneas antiestéticas, los resultados apuntan a un beneficio clínicamente relevante con mogamulizumab, en especial en pacientes con enfermedad avanzada y compromiso sanguíneo; los sujetos con enfermedad en estadios tempranos y sin afectación en sangre parecen experimentar menor beneficio. En base a ello, el IPT de la AEMPS establece que, frente al resto de alternativas, en el contexto de una MF o SS pre-tratada sería preferible el uso de esas alternativas en estadios iniciales y de mogamulizumab en estadios avanzados. En pacientes con patología CD30+ se podría valorar utilizar primero brentuximab vedotina, si bien la elección del tratamiento deberá hacerse de forma individualizada, considerando factores de la enfermedad y del paciente. En todo caso, mogamulizumab se usa con intención paliativa y no es una opción curativa, por lo que no parece representar una innovación terapéutica disruptiva.
Cáncer de pulmón no microcítico ALK+
Lorlatinib ▼LORVIQUA (Pfizer) PAM 441
Es un nuevo inhibidor selectivo, competitivo con el ATP y activo por vía oral, con actividad frente a las tirosina cinasas del linfoma anaplásico (ALK) y del oncogén c-ros 1 (ROS1). Las fusiones oncogenéticas de la ALK juegan un papel esencial en la regulación de la supervivencia de las células tumorales, el crecimiento y las metástasis, de modo que la inhibición de su actividad tirocina cinasa conllevará el bloqueo de las vías de señalización descendentes y la inducción de la muerte de la célula tumoral. En base a ello, el medicamento ha sido autorizado, en monoterapia, para el tratamiento de pacientes adultos con cáncer de pulmón no microcítico (CPNM) avanzado y positivo para la ALK cuya enfermedad ha progresado tras recibir alectinib o ceritinib como primer tratamiento con un inhibidor de la tirosina cinasa (ALK-TKI), o bien con crizotinib y al menos otro ALK-TKI.
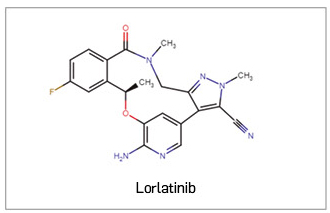
Los datos clínicos que han sustentado su autorización derivan de un ensayo pivotal no aleatorizado de fase 1/2, de un solo brazo, no aleatorizado ni controlado, que incluyó pacientes adultos con CPNM y afectación tumoral del SNC asintomática. En el conjunto de pacientes con ≥ 1 línea previa con un ALK-TKI de 2ª generación (N= 139), el tratamiento con lorlatinib mantenido hasta toxicidad inaceptable o progresión de la enfermedad indujo una TRO global del 39,6%, que a nivel intracraneal ascendió hasta el 56,1% (mayor que la respuesta extracraneal, del 36,7%). El efecto antitumoral fue rápido (mediana de tiempo hasta respuesta de 1,4 meses) y duradero (mediana de duración de la respuesta de 9,6 meses), lo que permitió alcanzar una supervivencia libre de progresión de 6,6 meses, siendo la mediana de supervivencia global de 20,7 meses. La eficacia del fármaco se mostró consistente e independiente de factores como la edad, el género, la raza o el estado funcional basal, e implicó una mejora significativa en la calidad de vida reportada por los pacientes. Con respecto a la seguridad, se trata de un fármaco relativamente bien tolerado, con un perfil de toxicidad manejable y en línea con los descritos para otros ALK-TKI. Se caracteriza por una alta incidencia eventos adversos, pero en su mayoría leves-moderados, entre los que destacan por su frecuencia: hipercolesterolemia (84%), hipertrigliceridemia (67%), edema (55%), neuropatía periférica (48%) y alteraciones cognitivas (29%). Entre los eventos adversos de grado ≥ 3 (39%) destacan la hipercolesterolemia (16%) y la hipertrigliceridemia (16%). Las reducciones de dosis fueron necesarias en un 23% de los pacientes, pero la tasa de discontinuación por eventos adversos asociados al fármaco fue baja (3%, sobre todo efectos cognitivos).
Así pues, es un nuevo inhibidor de ALK de 3ª generación que, por su amplio espectro de inhibición de formas mutantes de ALK y su capacidad de penetrar en el SNC, ha mostrado una eficacia antitumoral global e intracraneal clínicamente relevante: aporta un beneficio rápido, profundo y duradero en pacientes que han progresado tras un tratamiento con al menos un ALK-TKI de 2ª generación y que muestran metástasis cerebrales. Aunque con una estructura cíclica única entre los ALK-TKI, lorlatinib no aporta un grado de innovación destacable en el plano mecanístico ni supone una opción curativa. Se posicionará como una alternativa interesante y con un perfil favorable de toxicidad en 2ª línea o posteriores de farmacoterapia, en una necesidad médica no cubierta que hasta ahora no tenía un tratamiento estándar (ningún fármaco tiene autorizada exactamente la misma indicación, e incluso los ALK-TKI de 2ª generación difieren en sus condiciones de uso). Así, las principales guías clínicas europeas lo consideran como uno de los tratamientos de elección tras la progresión a un ALK-TKI de 2ª generación (alectinib o ceritinib). Hasta que se pueda concluir sobre el posible beneficio derivado de su uso en primera línea, lorlatinib solo va a beneficiar a una pequeña proporción de pacientes y no supondrá una modificación sustancial de la terapéutica estándar.
Brigatinib ▼ALUNBRIG (Takeda) PAM 445
Brigatinib es un nuevo inhibidor selectivo frente a las tirosina cinasas del linfoma anaplásico (ALK), del oncogén C-ROS 1 (ROS1) y del receptor 1 del factor insulínico de crecimiento (IGF-1R). Las fusiones oncogenéticas de la ALK juegan un papel importante en la regulación de la supervivencia de las células tumorales, el crecimiento del tumor y las metástasis, de modo que la inhibición de su actividad cinasa por brigatinib conllevará el bloqueo de las vías de señalización descendentes y la inducción de la muerte de la célula tumoral. En base a ello, el medicamento ha sido autorizado, en monoterapia, para el tratamiento por vía oral de pacientes adultos con cáncer de pulmón no microcítico (CPNM) avanzado, positivo para ALK, que no han sido pretratados con un inhibidor de ALK, o bien que han recibido previamente una línea de crizotinib.
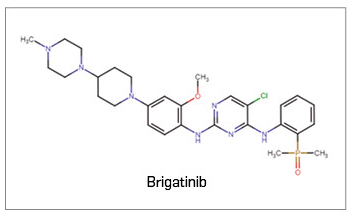
Los datos clínicos que sustentan su autorización en 2ª línea en pacientes que progresan o no toleran crizotinib derivan de un ensayo aleatorizado y abierto de fase 2 (N= 222), en que la pauta autorizada del fármaco ha demostrado una tasa de respuesta objetiva (TRO) del 56%, una mediana de supervivencia sin progresión (SLP) de 15,6 meses y una mediana de duración de la respuesta (DR) de 13,8 meses, mientras que los datos de supervivencia global (SG), no considerados maduros, sugieren una mediana de 34 meses; además, la TRO intracraneal alcanzó el 67% y la mediana de SLP intracraneal, los 18 meses. No se dispone de comparaciones directas con ceritinib ni alectinib en esta indicación y, aunque los resultados de eficacia con el nuevo fármaco parecen mejores, brigatinib puede considerarse por ahora una alternativa de tratamiento comparable a las anteriores y se requieren futuros datos para establecer la mejor secuencia terapéutica. Por otro lado, los datos de su uso en 1ª línea en pacientes sin tratamiento previo con un inhibidor de ALK proceden de un estudio pivotal abierto de fase 3 (N= 275), en que el tratamiento con brigatinib prolongó 13 meses la mediana de SLP frente a crizotinib (de 11 a 24 meses); también mejoró la TRO (74% vs. 62%), pero no se tienen datos maduros favorables al nuevo fármaco en términos de SG. Esa eficacia se corroboró a nivel del SNC, con una reducción del riesgo de progresión tumoral intracraneal del 70% y una multiplicación de la TRO (78% vs. 26%). En esta indicación tampoco se dispone de comparaciones directas frente a ceritinib o alectinib, si bien se ha postulado que brigatinib aportaría un beneficio similar a alectinib (considerado el tratamiento preferente en 1ª línea), y se posiciona como una opción preferente y alternativa a éste.
Con respecto a su seguridad, brigatinib tiene una toxicidad importante que puede afectar a la calidad de vida (más de la mitad de pacientes reporta algún evento adverso grave relacionado con el tratamiento), pero manejable clínicamente. Su perfil toxicológico es consistente tanto para su uso en primera como en segunda línea, y se caracteriza por efectos gastrointestinales, tos, aumento de CPK y enzimas hepáticas, cefalea, fatiga y disnea; es decir, está línea con lo descrito para otros inhibidores de ALK de 2ª generación. La hipertensión y los eventos pulmonares tempranos (enfermedad intersticial/neumonitis) parecen los aspectos exclusivos de brigatinib a valorar.
Así pues, se trata de un nuevo inhibidor de ALK de 2ª generación que, por su espectro de inhibición de formas mutantes de ALK y su capacidad de retrasar el crecimiento de las metástasis cerebrales (detectables en más de la mitad de la población diana), ha evidenciado una eficacia antitumoral global e intracraneal consistente en todos los subgrupos de pacientes y clínicamente relevante en un entorno de enfermedad no curable con pobre supervivencia global: es muy notable en pacientes pretratados con crizotinib y significativamente superior a ese fármaco en 1ª línea del tratamiento del CPNM ALK+ avanzado. No obstante, brigatinib no aporta novedad en el plano mecanístico ni incorpora ningún aspecto de mejora terapéutica sobre otras opciones disponibles, siendo sus posibilidades en la práctica clínica similares a las de otros fármacos ya autorizados.
Cáncer de próstata
Apalutamida ▼ERLEADA (Janssen-Cilag) PAM 441
Darolutamida ▼NUBEQA (Bayer) PAM 441
Apalutamida y darolutamida son dos nuevos inhibidores selectivos y potentes que bloquean varios pasos de la vía de señalización del receptor androgénico (RA): sin ejercer actividad agonista sobre el mismo y mediante su unión al dominio de unión de ligando, inhiben competitivamente la unión y activación por el andrógeno y, en última instancia, evitan la translocación al núcleo, la unión al ADN y la promoción de la transcripción génica por el RA. Así, reducen la proliferación y aumentan la apoptosis de las células tumorales de la próstata para conseguir un efecto antitumoral potente. En base a ello y a los datos mostrados en ensayos pivotales de fase 3 (aleatorizados, doble ciego y controlados por placebo), ambos fármacos han sido autorizados con indicación en hombres adultos para el tratamiento –por vía oral– del cáncer de próstata resistente a la castración no metastásico (CPRCnm) con alto riesgo de desarrollar metástasis; apalutamida se ha aprobado, además, frente al cáncer de próstata hormonosensible metastásico (CPHSm) en combinación con terapia de deprivación androgénica (TDA).
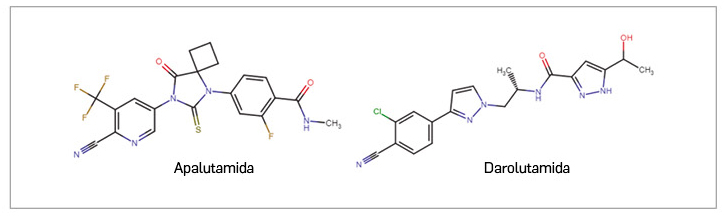
En un estudio con pacientes con CPRCnm y alto riesgo de metástasis y muerte por cáncer (N= 1.207), el tratamiento con apalutamida prolongó en más de 2 años –25 meses– la supervivencia libre de metástasis (SLM), reduciendo en un 70% el riesgo de metástasis a distancia (p< 0,0001). Aunque los resultados de supervivencia global (SG) no eran maduros ni significativos, se observó una tendencia favorable a apalutamida (mediana no alcanzada vs. 39,0 meses con placebo). Un segundo estudio con pacientes con CPHSm (N= 1.052) demostró una tendencia favorable en términos de SG (tasa de mortalidad de 16% vs. 22% con placebo; HR= 0,67; p= 0,0053) y de supervivencia sin progresión radiológica (HR= 0,48; p< 0,001); además, retrasó significativamente el inicio de la quimioterapia citotóxica (disminución de un 61% del riesgo de necesitarla en comparación con placebo). Por su parte, darolutamida fue superior a placebo en un estudio con pacientes adultos con CPRCnm y alto riesgo (N= 1.509), en que fue capaz de prolongar casi 2 años –22 meses– la SLM, reduciendo en un 59% el riesgo de metástasis o muerte (p< 0,0001). Si bien un análisis final tampoco pudo confirmar el efecto sobre la SG, la tendencia favorece a darolutamida, en cuyo brazo se registró una mortalidad menor (15,5% vs. 19,2% en el grupo placebo; HR= 0,69; p= 0,0030). Los resultados de otras variables secundarias respaldan la mejoría clínica con los dos nuevos fármacos. En relación a la seguridad, se trata de dos fármacos relativamente bien tolerados, definiéndose su perfil toxicológico por una incidencia global de eventos adversos similar a placebo. Como otros anti-andrógenos de su grupo, se caracterizan por reacciones adversas como fatiga, erupción cutánea o rash y dolores musculoesqueléticos. La gran mayoría de las relacionadas con el tratamiento son de una gravedad leve-moderada y manejables clínicamente, de modo que la proporción de pacientes que abandonó el tratamiento por motivos de seguridad fue similar a placebo, acaso solo levemente superior con apalutamida. En definitiva, es evidente que la adición de apalutamida o darolutamida a la terapia de deprivación androgénica (TDA, mantenida en todos los pacientes en ensayos clínicos) –castración quirúrgica previa o castración química con un análogo de GnRH– aporta un beneficio notable frente a la monitorización clínica con uso exclusivo de TDA en pacientes con cáncer de próstata resistente a la castración no metastásico de alto riesgo, al retrasar en aproximadamente 2 años la aparición de metástasis. A falta de resultados finales de los estudios en marcha, la relevancia de estos datos y su consistencia en todos los subgrupos de pacientes, superan las actuales incertidumbres relativas a la SG, la cual, igual que la calidad de vida reportada por los pacientes, no se ve perjudicada. Hasta que se disponga de comparaciones directas frente a enzalutamida, parece que estos nuevos fármacos se incorporan como alternativas de tratamiento en el mismo grupo terapéutico, sin representar una innovación farmacológica o clínica disruptiva, desconociéndose su perfil beneficio-riesgo en pacientes con un estado funcional inicial más degradado (ECOG > 1).
Esclerosis múltiple
Siponimod ▼MAYZENT (Novartis) PAM 445
Siponimod es un modulador del receptor de la esfingosina 1-fosfato (S1P) que se une selectivamente a los receptores S1P1 y S1P5. Al actuar como un antagonista funcional en los receptores S1P1 de los linfocitos, que se insensibilizan de forma dosis-dependiente al efecto de la S1P, el fármaco previene la salida de dichas células desde los órganos linfoides y, en consecuencia, provoca una redistribución linfocitaria: quedan “secuestrados” y se reduce su infiltración patológica al sistema nervioso central, limitando la inflamación y lesiones en el tejido nervioso; además, siponimod reduce el recuento de linfocitos periféricos en un 20-30% respecto a niveles basales. En base a ello, el medicamento ha sido autorizado para el tratamiento por vía oral de pacientes adultos con esclerosis múltiple secundaria progresiva (EMSP) con enfermedad activa definida por brotes o por características de imagen típicas de actividad inflamatoria.
Los datos clínicos que han sustentado su autorización derivan de un único ensayo pivotal multicéntrico de fase 3 con una fase principal doble ciego y controlada, en que el tratamiento diario con siponimod demostró superioridad clínica sobre placebo en pacientes con EMSP (N= 1.651). En esa comparativa, se observó un beneficio relevante con el uso del fármaco en la población global de pacientes, plasmado en el retraso de la progresión de la discapacidad, con una reducción relativa –estadísticamente significativa– del riesgo del 21% a los 3 meses y del 26% a los 6 meses, así como en una reducción del 55% en la tasa anualizada de brotes. La superioridad del fármaco sobre placebo, consistente en todos los subgrupos analizados, fue mayor en los pacientes con enfermedad activa al inicio, en quienes redujo el riesgo de progresión de la discapacidad en un 31% y 37% a los 3 y 6 meses, respectivamente, disminuyendo en un 46% la tasa de brotes confirmados. En todo caso, la baja proporción de pacientes que progresó tras 3 años de seguimiento, la ausencia general de significación estadística en los resultados de calidad de vida reportados por los pacientes o la inclusión de pacientes con enfermedad y progresión moderada (impide probar convincentemente la prevención de la progresión de la discapacidad) contribuyen a cuestionar la relevancia clínica de los resultados. Con respecto a la seguridad, siponimod muestra un perfil toxicológico complejo, pero similar al del otro miembro de su grupo farmacológico –fingolimod–, que se asocia con una baja tasa de discontinuaciones.
La incidencia de eventos adversos es escasamente superior a placebo, pero sobresalen riesgos de seguridad relativos a la frecuencia de infecciones y a los eventos adversos cardiovasculares (bradicardia o bloqueo de la conducción cardiaca), que hacen necesario un seguimiento específico de los pacientes. Por su frecuencia destacan las alteraciones del sistema nervioso, cefalea e hipertensión, mayoritariamente leves-moderadas, mientras que por su severidad se subraya el riesgo de infecciones del tracto urinario, elevaciones de ALT y depresión.
Este nuevo fármaco modificador de la enfermedad aporta un beneficio clínico superior a placebo en pacientes con EMSP y continúa la vía terapéutica inaugurada por fingolimod, sin aportar innovación a nivel mecanístico. El IPT lo considera una alternativa a interferón-β 1b, ocrelizumab o cladribina en el tratamiento de pacientes con EMSP y actividad patológica, sin ser posible establecer la superioridad de uno sobre otro; aunque la evidencia en el caso de siponimod es más robusta que para ocrelizumab o cladribina, no se puede concluir sobre su beneficio en todo el espectro de la enfermedad con EMSP. Tampoco aporta ningún otro aspecto innovador reseñable (vía de administración novedosa en la indicación o mejor perfil toxicológico).
Artritis reumatoide
Filgotinib ▼JYSELECA (Gilead) PAM 449
Filgotinib es un nuevo inmunosupresor activo por vía oral que actúa mediante la inhibición selectiva, reversible y competitiva con el ATP, de las cinasas Janus (JAK), preferentemente sobre las JAK-1/JAK-3, de manera que modula la respuesta inflamatoria e inmunitaria mediada por citocinas o factores de crecimiento. Estrechamente relacionado con otros inhibidores de JAK previamente comercializados en España (tofacitinib, baricitinib y upadacitinib), el medicamento ha sido autorizado para el tratamiento por vía oral –tanto en monoterapia como asociado a metotrexato– de la artritis reumatoide (AR) activa de moderada a grave en pacientes adultos con respuesta inadecuada o intolerancia a uno o más fármacos antirreumáticos modificadores de la enfermedad (FAME).
Su eficacia ha sido adecuadamente contrastada en 3 ensayos clínicos pivotales de fase 3, controlados y doblemente ciegos, que han aleatorizado más de 3.400 pacientes, y en los que ha demostrado una superioridad estadísticamente significativa frente a placebo en pacientes con fracaso a FAME convencionales y/o biológicos, y también mayor que metotrexato (en pacientes naïve para su uso o con respuesta inadecuada a dicho FAME) y adalimumab (en pacientes con fracaso previo a metotrexato). Tanto en segunda como en tercera línea de tratamiento, y tanto en monoterapia como en combinación con FAME convencionales, la dosis de 200 mg/día de filgotinib permitió alcanzar a los 3 meses tasas de respuesta según criterios ACR20 del 66-77% (vs. 31-50% con los comparadores), lo que se traducía en un aumento notable de la proporción de pacientes en remisión clínica según la puntuación de DAS28-PCR (22-34% vs. 8-9%); se confirmó la no-inferioridad del fármaco frente a adalimumab. Con una eficacia de inicio rápido (evidente desde la semana 2) y duradera (mantenida en periodos de 1 año e incluso creciente en tratamientos de hasta 4 años en estudios de extensión), se confirmó el beneficio de su uso en todos los subgrupos de pacientes, que se completó con mejoras en variables secundarias como la progresión radiológica de la enfermedad, la función física de los pacientes o la calidad de vida. Por otro lado, el perfil toxicológico del fármaco, similar en su uso en monoterapia y en combinación con FAME convencionales, es manejable clínicamente y concuerda con los riesgos ya conocidos de los fármacos inhibidores de las JAKs. Entre los eventos adversos asociados al tratamiento destacan por su frecuencia las infecciones –especialmente del tracto respiratorio superior y del tracto urinario–, y otras reacciones adversas como náuseas, mareos y ciertas alteraciones analíticas. La tasa de mortalidad no difiere sustancialmente de la de los comparadores activos empleados ni tampoco si se compara indirectamente con la de otros inhibidores de JAK. Así, el seguimiento que requieren los pacientes tratados con filgotinib es similar a la monitorización común en el abordaje de la AR.
No se dispone de comparaciones directas frente a otros tratamientos de los que podría ser una alternativa (con excepción de adalimumab) y la evidencia derivada de comparaciones indirectas, no concluyente, sugiere que filgotinib no es superior a otros fármacos de su grupo. En definitiva, a falta de conocer las consideraciones del IPT, se puede posicionar como una alternativa válida más dentro de los FAME sintéticos dirigidos, similar a los otros inhibidores de JAK autorizados, entre los cuales todavía no se puede establecer diferencias sustanciales, y sin aportar ninguna mejora aparente respecto a éstos. Los usos autorizados constituyen una 2ª o 3ª línea de tratamiento y no van a suponer una modificación de la terapéutica estándar de la artritis reumatoide.
Sistema nervioso
Narcolepsia
Pitolisant ▼WAKIX (Bioprojet Pharma) PAM 440
Pitolisant es un potente antagonista/agonista inverso de los receptores H3 de la histamina que, mediante el bloqueo de dichos autorreceptores, aumenta la actividad de las neuronas histaminérgicas; también potencia la liberación de acetilcolina, noradrenalina y dopamina. Puesto que la pérdida de neuronas secretoras de hipocretina en la narcolepsia se puede compensar por la activación de neuronas histaminérgicas –situadas mayoritariamente en el hipotálamo posterior, pero con proyecciones por todo el cerebro–, la acción de pitolisant sobre los receptores H3 ejerce una potenciación prolongada de la transmisión histaminérgica capaz de aumentar el estado de alerta y vigilia. En base a ello, el medicamento, designado como huérfano y de administración por vía oral, ha sido autorizado para el tratamiento de la narcolepsia con o sin cataplejía en adultos.
La eficacia del fármaco en la indicación y dosis autorizadas ha sido contrastada mediante dos ensayos pivotales multicéntricos de fase 3 y con un diseño adecuado: aleatorizados, doble ciego, de grupos paralelos y controlados por placebo y modafinilo, de 8 semanas duración. En uno de ellos, pitolisant mostró superioridad sobre placebo en términos de reducción de la somnolencia diurna excesiva, con una diferencia media –tras ajuste a valores basales– de -3,3 puntos (p< 0,05) en la escala ESS. No alcanzó, en cambio, significación estadística en el otro ensayo (diferencia de solo -1,9 puntos; p= 0,065), posiblemente por la diferencia en las dosis estudiadas. Los resultados de las variables secundarias (por ejemplo, la tasa de respondedores según la puntuación ESS o la capacidad de mantener la vigilia según la escala MWT) respaldan su superioridad sobre placebo. Además, los hallazgos de un estudio de soporte reafirman su beneficio sobre la cataplejía: reduce a más de la mitad la tasa de episodios semanales tras 4 semanas de tratamiento, desde 9,2 hasta 3,3 (vs. una reducción de 7,3 hasta 6,8 con placebo; p< 0,0001). Sin embargo, los ensayos pivotales no pudieron demostrar la no-inferioridad de pitolisant frente a modafinilo: en uno de los estudios, incluso, la diferencia entre sus efectos sobre la escala ESS fue de -2,75 puntos a favor de modafinilo, que se mostró más eficaz que pitolisant (p< 0,002). Pero hay que subrayar que, a diferencia de modafinilo, pitolisant sí ha demostrado actividad sobre la cataplejía. En términos de seguridad, pitolisant exhibe una buena tolerabilidad: la proporción de pacientes tratados que reporta algún evento adverso (52%, la mayoría de casos leves a moderados) es ligeramente superior a la de placebo (41%) y similar a la de modafinilo (55%). La cefalea (11%) y el insomnio (9%) son las reacciones adversas más frecuentes y las que provocan más retiradas del tratamiento, aunque también destacan los trastornos gastrointestinales –náuseas, vómitos o diarrea–, psiquiátricos –ansiedad, irritabilidad o depresión– y aumento de peso. Con un riesgo no desdeñable de interacciones farmacológicas, pitolisant presenta un perfil de seguridad diferente al de modafinilo (menor gravedad, menor incidencia de algunas reacciones adversas y un aparente menor potencial de dependencia) y al de oxibato (el cual tiene un alto potencial de abuso y posibles reacciones adversas graves).
En definitiva, ha demostrado una eficacia superior a placebo en pacientes adultos con narcolepsia, siendo capaz de reducir significativamente la somnolencia diurna excesiva y la incidencia de la cataplejía, pero aún persisten ciertas dudas sobre la magnitud del beneficio clínico, sobre todo, por la limitación de los datos clínicos a largo plazo (en una enfermedad crónica que requerirá tratamientos prolongados). Así, puede considerarse por el momento como una nueva alternativa terapéutica a modafinilo u oxibato sódico en segunda línea, teniendo en cuenta las características individuales del paciente, su perfil de seguridad y posología (más cómoda que oxibato). Inaugura un mecanismo de acción en una enfermedad rara, pero representa solo un tratamiento sintomático que no va a variar sustancialmente la terapéutica. Los estudios poscomercialización y la farmacovigilancia serán esenciales para caracterizar en profundidad el balance beneficio-riesgo del nuevo fármaco a largo plazo y esclarecer su prometedor papel como una potencial primera línea en el tratamiento de la narcolepsia.
Órganos de los sentidos
Distrofia retiniana por mutación RPE65
Voretigén neparvovec ▼LUXTURNA (Novartis) PAM 446
Es un vector recombinante de transferencia genética basado en un adenovirus de serotipo 2 que introduce, cuando se administra por inyección subretiniana, el ADN complementario de la proteína RPE65 humana en la retina. Con la transducción de las células epiteliales del pigmento retiniano, proporciona el potencial necesario para que se regenere a nivel intraocular el 11-cis-retinal y se restaure el ciclo visual en pacientes que tienen ausencia o déficit de la actividad enzimática todo-transretinil isomerasa de la RPE65. En base a ello, este medicamento de terapia génica, designado como huérfano, ha sido autorizado para el tratamiento –en dosis única en cada ojo– de adultos y niños con pérdida de visión debido a una distrofia retiniana hereditaria asociada a la mutación RPE65 bialélica confirmada y que tienen suficientes células retinianas viables.
Los datos clínicos conducentes a su aprobación derivan fundamentalmente de un ensayo pivotal abierto de fase 3, aleatorizado y controlado en el que participaron 31 pacientes de ≥ 3 años con amaurosis congénita de ACL debido a mutaciones de RPE6517. Tras 1 año desde la administración, la diferencia media en la puntuación de la prueba de movilidad multiluminiscente bilateral entre el grupo tratado y el control fue de +1,6 puntos con la visión binocular (p< 0,001); el 62% de los pacientes completó la prueba a 1 lux como iluminación mínima (vs. ninguno en el grupo control). Una magnitud similar del cambio (de 1,7-2,0 puntos) se observó también con la visión monocular. Los pacientes del grupo control, que al año no mostraron ninguna mejoría en su función visual, tuvieron un beneficio similar cuando se cruzaron a recibir el medicamento. Otras variables secundarias, como el test de sensibilidad a la luz de campo completo (diferencia media tras 1 año de -2,11 puntos como promedio de ambos ojos, a favor de voretigén neparvovec) o la evaluación de la agudeza visual (cambio significativo en el 55% de los pacientes en el primer ojo y en el 20% en el segundo, frente a ninguno en el grupo control), corroboraron la mejora de la función visual con la terapia experimental, la cual fue evidente desde 1 mes postratamiento, y se mantiene durante al menos 3 años.
Con respecto a la seguridad, es un medicamento bien tolerado. Pueden aparecer alteraciones visuales (visión borrosa o fotofobia) durante algunas semanas, pero la gran mayoría de eventos adversos notificados fueron leves, transitorios y no relacionados con el vector. Más de la mitad de pacientes refiere algún trastorno ocular, gastrointestinal o del sistema nervioso, destacando la incidencia de cefalea (45%), leucocitosis (38%), náuseas (35%) y vómitos (35%). Hubo 3 casos de depósito retiniano en un solo ojo probablemente relacionados con el fármaco, pero fueron asintomáticos, sin correlación clínica y autolimitados. Además, es probable una alta frecuencia de reacciones adversas relacionadas con la administración (69%, graves solo el 7%); las más frecuentes son: hiperemia conjuntival, cataratas, aumento de la presión intraocular, dolor ocular o desgarro retiniano. No obstante, habrá que esperar a los resultados de estudios posautorización, con periodos de seguimiento de hasta 15 años, para caracterizar en mayor medida el balance beneficio-riesgo a largo plazo.
Se trata de una nueva terapia génica que representa una innovación farmacológica disruptiva por inaugurar una vía terapéutica en términos de mecanismo de acción y por aportar un beneficio clínicamente relevante en pacientes, en su mayoría niños o adolescentes, que hasta ahora no tenían ninguna opción de tratamiento autorizado en Europa para mejorar o recuperar su visión, abriendo incluso una puerta de esperanza a su curación con el progreso científico. Por ser el primer medicamento aprobado en su indicación, no puede realizarse ninguna comparación directa o indirecta con otra alternativa farmacológica, y se posicionará como elección preferente en primera línea. No obstante, se identifican algunos factores que pueden limitar su potencial uso o valor terapéutico: se restringe a la administración por cirujanos experimentados en cirugía macular y la proporción de pacientes que puede beneficiarse del tratamiento es baja (solo un 10% de todos los casos de amaurosis congénita de Leber y el 3% de los pacientes con retinosis pigmentaria presentan mutaciones bialélicas en el gen RPE65).
Varios
Alergia al veneno de himenópteros
Alérgenos de veneno de abeja ▼ALLUTARD SQ APIS MELLIFERA (ALK Abelló) PAM 444
Alérgenos de veneno de avispa ▼ALLUTARD SQ VESPULA SPP (ALK Abelló) PAM 444
Estamos ante dos medicamentos a base de extractos estandarizados de alérgenos de veneno de abeja y avispa, respectivamente, que representan una nueva inmunoterapia desensibilizante para reducir los síntomas de la respuesta inmunitaria, actuando sobre los mecanismos humorales y celulares de la inflamación alérgica: median la modulación de la respuesta de anticuerpos alér¬geno-específicos, la disminución en el reclutamiento y activación de células proinflamatorias y los cambios en el patrón de respuesta de las células T alérgeno-específicas (hacia Th1). En base a ello, los medicamentos han sido autorizado para su administración por vía subcutánea en pacientes con historia clínica documentada de reacciones alérgicas generalizadas y/o sistémicas mediadas por IgE –confirmada por prueba cutánea de prick, prueba intradérmica o prueba IgE específica– debidas a la sensibilización al veneno de abeja (Apis mellifera) y de avispa (Vespula spp.), respectivamente.
La potencial amenaza vital que representan esas reacciones alérgicas y otros motivos éticos han impedido la realización de estudios específicos controlados por placebo. Su autorización se ha visto sustentada por la literatura científica derivada de estudios clínicos de hasta 5 años de duración, que, en tratamientos de hasta 1 año con el veneno de abeja y hasta 3 años con veneno de avispa, han permitido contrastar tasas de eficacia protectora consistentemente superiores al 80%; o sea, esa amplia proporción de pacientes no sufrió una reacción alérgica sistémica o generalizada como consecuencia de una picadura, bien fortuita en el medio natural o bien tras una prueba hospitalaria de re-picadura. Los medicamentos presentan, además, una relativa buena tolerabilidad, gracias sobre todo a la pauta de administración con dosis crecientes de alérgeno. El perfil toxicológico se caracteriza por eventos adversos asociados a la respuesta alérgica al veneno, que pueden manejarse con la administración de antihistamínicos. Aunque la incidencia de los eventos adversos locales es variable según los estudios (6-79% en la fase de inicio y 0-47% en mantenimiento), los mayoritarios son las reacciones inmediatas en el lugar de inyección (urticaria, dolor, eritema, hinchazón, prurito, etc.). Entre las reacciones adversas sistémicas se describen prurito ocular, rinitis, asma y tos, urticaria y angioedema; son menos frecuentes (0-25% en la fase de inicio y 0-16% en mantenimiento) y en su mayoría leves-moderadas, si bien por su severidad sobresale el riesgo de shock anafiláctico, que requeriría tratamiento inmediato con adrenalina.
En vista de lo anterior, los dos nuevos medicamentos no suponen una novedad excepcional en el tratamiento de enfermedades alérgicas desde el punto de vista del mecanismo farmacológico, pero aportan un grado notable de innovación terapéutica por ser las primeras opciones de inmunoterapia desensibilizante para pacientes con alergia debidamente contrastada al veneno de abejas y avispas que sufran reacciones sistémicas o generalizadas (se ha descrito una prevalencia de reacciones alérgicas al veneno de estos insectos en torno al 3% de la población en Europa). Se debe subrayar que no se indican en pacientes con reacciones locales (la mayoría de las personas), aunque sean intensas, ni en reacciones no-mediadas por IgE. Si bien la ruta de administración subcutánea puede conllevar ciertos inconvenientes en términos de adherencia terapéutica (frente a la vía oral o sublingual), la formulación depot –adsorción en hidróxido de aluminio– de los dos medicamentos representa una ventaja potencial, al asociarse con una mejor tolerabilidad local que los extractos acuosos de veneno de himenópteros.
Hiperpotasemia
Ciclosilicato de sodio y circonio ▼LOKELMA (AstraZeneca) PAM 445
El ciclosilicato de sodio y zirconio (zirconium ciclosilicato, ZC) es un compuesto no polimérico y no absorbible con una estructura de microporos que tiene una alta selectividad hacia los iones potasio (incluso en presencia de otros cationes): captura dichos iones K+ a lo largo de todo el tracto gastrointestinal y los intercambia por cationes sodio e hidrógeno, reduciendo la concentración de potasio libre y aumentando su excreción a nivel fecal. Así, el fármaco reduce los niveles séricos de K+ rápidamente tras su administración oral, de modo que el medicamento ha sido autorizado para el tratamiento de la hiperpotasemia en adultos.
La eficacia clínica del fármaco se ha confirmado fundamentalmente en 3 ensayos pivotales aleatorizados y doble ciego que han incluido pacientes adultos con hiperpotasemia leve a moderada, mayoritariamente con niveles de K+ séricos < 5,5 mmol/l, con enfermedad renal crónica, uso concomitante de fármacos inhibidores del sistema renina-angiotensina-aldosterona (por ejemplo, IECAs o ARA-II) y/o diabetes mellitus. Se ha probado su superioridad frente a placebo en un tratamiento de corrección de 48-72 h –fase aguda–, en que aumenta significativamente la proporción de pacientes con normopotasemia (80-86% con la dosis de 10 g/3 veces al día vs. 48% con placebo), y también en tratamientos de mantenimiento –fase subaguda– de entre 12 y 28 días y hasta 1 año de duración, en que incrementa notablemente la proporción de pacientes que continúan en normopotasemia (82-94% a la dosis de 10 g/día vs. 46-57% con placebo) y el número de días en normopotasemia, y reduce los niveles promedio de K+ sérico. Un estudio adicional demostró que el tratamiento con ZC durante 8 semanas también aumenta significativamente la proporción de respondedores con normopotasemia frente a placebo entre los pacientes que requieren hemodiálisis (41% vs. 1%). Por otra parte, se trata de un fármaco bien tolerado, con un perfil toxicológico esperable y similar al de las resinas de intercambio catiónico y a patiromero. La incidencia de eventos adversos baja (< 35%) y similar a placebo con las dosis autorizadas, tanto en tratamiento a corto como a más largo plazo, predominando los eventos adversos gastrointestinales (diarrea, náuseas, vómitos, estreñimiento y dolor abdominal), en su amplia mayoría leves-moderados; por su especial interés se notificaron hipopotasemia (4%) y eventos relacionados con edemas (6%), manejables clínicamente. Solo una escasa proporción se consideraron graves (4,2% vs. 1,7% con placebo), sin relación con la dosis recibida. El fármaco carece de los riesgos de hipernatremia o hipo/hipercalcemia relacionados con las resinas sódica y cálcica de intercambio catiónico, o la hipomagenesemia asociada a patiromero.
A fin de posicionar el fármaco en el arsenal terapéutico, se debe subrayar que en el tratamiento de la hiperpotasema leve-moderada a corto plazo no existen datos de eficacia y seguridad frente a las resinas intercambiadoras de cationes, las cuales también se han probado superiores a placebo (aunque con evidencia muy limitada): en ese contexto, ZC no parece aportar ventajas adicionales –tampoco en términos de vía de administración o pauta posológica– y podría considerarse como una nueva alternativa de tratamiento a las resinas similar en eficacia. En el tratamiento de la hiperpotasemia leve-moderada a largo plazo tampoco existen datos comparativos directos entre ZC y patiromero; ambos han demostrado una capacidad significativamente superior a placebo en el control del K+ plasmático, de modo que ZC también podría considerarse una alternativa de tratamiento a patiromero en ese escenario clínico. En definitiva, no parece que el nuevo fármaco suponga una innovación notable, ni en términos mecanísticos ni terapéuticos, debiendo tenerse en cuenta que los datos clínicos en pacientes con hiperpotasemias moderadas son muy limitados y casi nulos para las hiperpotasemias graves.