Resumen
Daridorexant es un novedoso antagonista del receptor de la orexina, que ejerce una acción dual y equipotente sobre los receptores orexina 1 y orexina 2, bloqueando el efecto promotor de la vigilia que los neuropéptidos orexina A y orexina B ocasionan al actuar sobre ellos. Se diferencia, pues, de otros fármacos hipnóticos en que no provoca una inhibición generalizada de la actividad cerebral. Por su capacidad de evitar la activación de los receptores de la orexina y reducir el desencadenante de la vigilia, facilita el sueño sin alterar su estructura normal, siendo esta la base de la autorización del medicamento para el tratamiento por vía oral de pacientes adultos con insomnio caracterizado por presencia de síntomas durante al menos 3 meses e impacto considerable en la actividad diurna.
La eficacia del nuevo fármaco se ha contrastado en dos estudios pivotales de similar diseño (doble ciego, multicéntrico, aleatorizado y controlado por placebo). En ellos la pauta recomendada (50 mg/día) ha probado una eficacia significativamente superior frente a placebo en variables objetivas y subjetivas que reflejan convenientemente la severidad del insomnio y su impacto en la funcionalidad diurna. Así, tras un mes de tratamiento, daridorexant redujo significativamente el tiempo de latencia del sueño –que al inicio rondaba 1 h de media– entre 6 y hasta 12 min frente a placebo, con una magnitud del efecto constante tras 3 meses. También en comparación con placebo, el tiempo despierto tras el inicio del sueño –que en el estado basal superaba la hora y media– se vio reducido en un promedio entre 12 y 23 min, con un efecto sostenido hasta el mes 3. Estos resultados fueron consistentes en los subgrupos evaluables de pacientes, con independencia de factores como la edad, el sexo o la raza.
El perfil de seguridad de daridorexant es aparentemente benigno. La frecuencia de eventos adversos en los dos estudios fue comparable entre ambos brazos de tratamiento: 38% con daridorexant 50 mg/día, 38-39% con daridorexant 25 mg/día y 33-34% con placebo. Los eventos adversos más frecuentes fueron cefalea (4-6% vs. 4% con placebo), somnolencia (2-3% vs. 2%) y nasofaringitis. Este perfil de seguridad parece mantenerse constante durante al menos 1 año y también es comparable entre grupos etarios.
Daridorexant es, por tanto, un innovador fármaco que inaugura una vía terapéutica en su indicación. Con un inicio rápido de su efecto farmacológico, reduce el exceso de alerta en la vigilia, habiéndose probado superior a placebo en la inducción y el mantenimiento del sueño, sin afectar a la estructura del mismo ni a la capacidad funcional de la persona al día siguiente. Constituye la primera incorporación al arsenal terapéutico del insomnio en más de dos décadas, y el primero que aborda de manera específica la fisiopatología y ha ganado indicación en insomnio crónico, por ser compatible con un uso a largo plazo (aunque lo ideal es la mínima duración del tratamiento, que debe reevaluarse cada 3 meses). Se prevé, por tanto, que pueda suponer una modificación importante de la terapéutica estándar en pacientes con insomnio crónico (entre el 8% y el 12% de la población). En cualquier caso, su eficacia parece moderada y hay que recordar que se posicionará como una segunda línea de tratamiento tras la intervenciones cognitivo-conductuales.
Aspectos fisiopatológicos
El insomnio se define, según la ICSD-31, como una dificultad persistente en el inicio del sueño, su duración, consolidación y/o calidad que ocurre a pesar de la existencia de adecuadas circunstancias y oportunidad para el mismo, acompañándose de un nivel significativo de malestar o deterioro de varias áreas de la vida del individuo. En otras palabras, en el insomnio es habitual la incapacidad para conciliar el sueño, el aumento del número de despertares nocturnos o la disminución del tiempo de sueño (aunque la menor duración del sueño por sí sola no define el insomnio) con despertares demasiado tempranos, e implica una sensación subjetiva negativa respecto a la duración o la calidad del mismo, percibido como insuficiente y no reparador.
Es importante considerar que el efecto de este trastorno no se limita a las horas nocturnas, ya que afecta a la actividad cotidiana diurna del sujeto y a su capacidad para recuperar el tono normal de funcionamiento vital, con una disminución de la atención, un menor rendimiento intelectual, un descenso notable de la energía y una mayor irritabilidad e inestabilidad emocional, entre otras posibles manifestaciones. En niños, el insomnio suele ser comunicado por sus padres o cuidadores y se manifiesta por resistencia activa para irse a la cama por las noches, despertares frecuentes durante la noche e incapacidad para dormir solo.
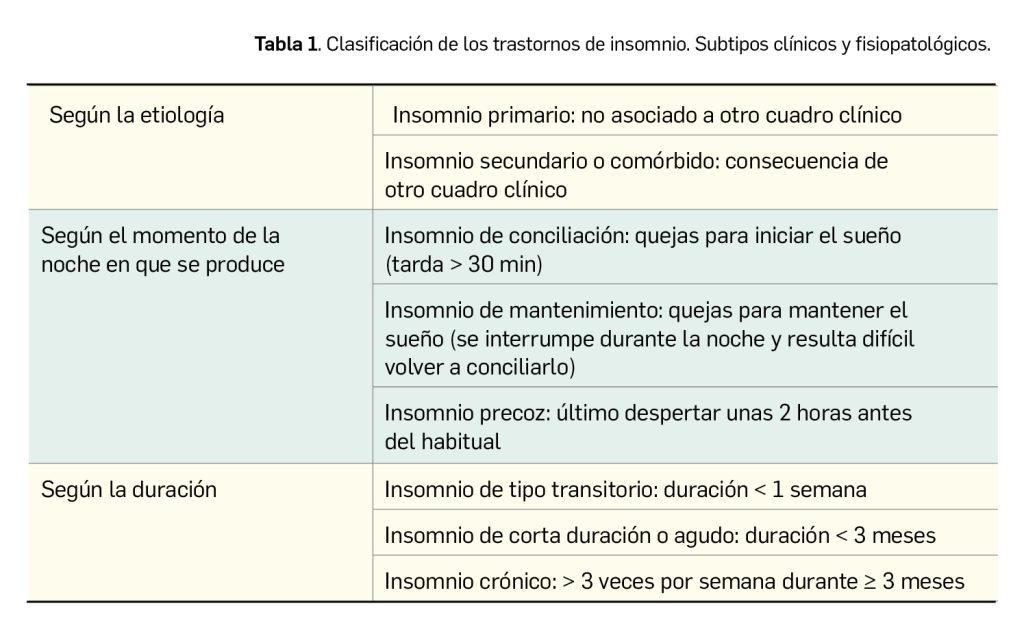
Además de que, por su intensidad, se suele diferenciar entre leve, moderado o grave, la clasificación ICSD-3 identifica varios tipos de insomnio en función de la etiología, del momento en que se produce y, sobre todo, de la duración las dificultades en el inicio o el mantenimiento del sueño con síntomas asociados durante el día (Tabla 1). El diagnóstico de esta enfermedad es eminentemente clínico, en base a los citados criterios diagnósticos o a los del DSM-V (Diagnostic and Statistical Manual of Mental Disorders, 5ª edición) de la Asociación Americana de Psiquiatría.
Como su clasificación, la etiología del insomnio es muy diversa: en la mayoría de los casos (≈80%) puede ser consecuencia de trastornos orgánicos (por ejemplo, hipertiroidismo o patologías que producen dolor), psíquicos o psiquiátricos (por ejemplo, ansiedad), o de factores externos (en cuyo caso se considera insomnio secundario), si bien hasta en un 20% de pacientes se puede considerar como un trastorno primario. Para confirmar este último caso, las manifestaciones se deben presentar al menos 3 veces a la semana durante al menos 1 mes. No obstante, es difícil en muchas ocasiones determinar si un trastorno externo provoca insomnio o, en realidad, dicho trastorno es debido al insomnio primario; es decir, la relación causa-efecto en estos casos es ambigua. Es más, el insomnio es considerado como un factor de riesgo para la mayoría de las patologías con las que coexiste, incluyendo la enfermedad coronaria y la depresión; por este motivo, tiende a utilizarse el término insomnio comórbido preferentemente al de insomnio secundario.
Habitualmente considerado como un trastorno del sueño “clásico”, la fisiopatología del insomnio sugiere un fenómeno más complejo: por un lado, una combinación de un estado de hiperexcitación psicofisiológica durante el sueño y de exceso de alerta (frente al perfil del sueño fisiológico), con aumento de la tasa metabólica por una mayor actividad cerebral, y mayores niveles de cortisol y de corticotropina (ACTH) durante la fase inicial del sueño en estos pacientes; y, por otro, una reducción del tono parasimpático en el proceso de adaptación de la respuesta cardiaca y un aumento de la actividad electroencefalográfica de alta frecuencia durante el sueño no REM. En resumen, se ha descrito una hiperactividad del sistema de respuesta de estrés (CRH-ACTH-cortisol y simpático) y alteraciones en el ritmo de secreción de las citocinas proin-flamatorias, tales como IL-6 y TNF-α (Buysse, 2013). En los últimos años ha ganado relevancia el papel que en la fisiopatología del insomnio juegan las orexinas, unos neuropéptidos secretados en el hipotálamo lateral que tienen como función el mantenimiento de la vigilia, y que están deficitarios, por ejemplo, en personas con narcolepsia.
En términos epidemiológicos se estima que hasta un tercio de la población puede sufrir insomnio, siendo más prevalente en mujeres y en personas con bajo nivel socioeconómico. La amplia variabilidad de las cifras de prevalencia entre distintos estudios parece deberse a diferencias metodológicas en cuanto a la definición de insomnio aplicada; por ejemplo, se ha observado que no todas las quejas de insomnio conllevan sintomatología diurna asociada, cuya consideración hace disminuir las cifras de prevalencia hasta el 10%. Se ha postulado que las manifestaciones del insomnio pueden tener una prevalencia anual del 35-50% entre la población general adulta, mientras que la del insomnio propiamente dicho estaría en el rango del 3-12% en adolescentes y 15-20% en adultos (crónico en el 6-12% de la población), creciendo notablemente hasta el 30% en mayores de 55 años. De hecho, por el deterioro de la calidad del sueño que se produce con la edad (cambios fisiológicos2), el incremento de la comorbilidad y la polimedicación, el insomnio alcanza su mayor prevalencia en la población anciana (Molina et al., 2019).
En España, se calculó que en torno a un 21% de las personas mayores de 15 años tienen insomnio, siendo más frecuente en las mujeres que en los varones (24% vs. 18%) e incrementándose la prevalencia con la edad. Otro estudio realizado en Canadá encontró que el tipo de insomnio relacionado con un acortamiento sustancial del sueño era la forma más común (50-70%), seguido de las dificultades para conciliarlo (35-60%) y el carácter no restaurador del sueño (20-25%); muchos pacientes referían dos o más tipos diferentes de insomnio. Además, se estima que el insomnio tiende a hacerse crónico en una amplia proporción (40-70%) de los individuos que dicen padecerlo.
Por lo general, se acepta que una serie de factores de riesgo se asocian a una mayor prevalencia de insomnio, tales como: la coexistencia de depresión, sexo femenino, edad avanzada, bajo estatus económico, patologías crónicas concomitantes, estado marital (mayor riesgo para divorciados o separados que para quienes viven en pareja o han permanecido siempre solteros) y raza (mayor riesgo para los afroamericanos que para los caucásicos). Asimismo, como ya se ha sugerido, el propio insomnio se considera un factor de riesgo para las enfermedades psiquiátricas –en particular, para la depresión (aumenta el riesgo a más del doble que en población general) y para el riesgo de peor respuesta a su tratamiento–, el síndrome metabólico, la hipertensión y la enfermedad coronaria. También es importante tener en cuenta lo que se conoce como factores perpetuadores, que tienden a convertir el insomnio en un círculo vicioso de causa-efecto-causa: un ejemplo característico es el comportamiento de muchos individuos insomnes que se mantienen en la cama durante periodos muy prolongados, con la esperanza de que el sueño acabe por venir, cuando, en realidad, esta prolongación de la estancia en cama tiende más bien a prolongar el insomnio y aumentar la ansiedad asociada al mismo (Fernández-Moriano, 2021).
A fin de mejorar la calidad y cantidad del sueño (para contrarrestar el impacto sobre la vida diaria y la salud de la persona afectada), la primera línea del tratamiento y de la prevención del insomnio se fundamenta en medidas no farmacológicas, entre las que indudablemente destaca la educación sobre la higiene del sueño. Asimismo, la terapia cognitivo-conductual (TCC)3, respaldada por una amplia evidencia, es la técnica psicoterapéutica más empleada en el abordaje del insomnio. Busca modular los factores que provocan la persistencia y cronificación del trastorno, y sus efectos a corto o medio plazo (6-10 semanas) son comparables o incluso superiores a los conseguidos con fármacos y persisten durante al menos 3 años. La TCC es también eficaz en pacientes que están tomando hipnóticos e incluso puede ayudar a reducir la utilización de estos últimos. También pueden ser útiles otras psicoterapias, como el control de estímulos (basada en fortalecer la relación cama/dormitorio con relajación y sueño), la restricción del sueño (limita el tiempo que el paciente pasa en la cama cada noche), los ejercicios de relajación o la terapia cognitiva de restructuración de pensamientos erróneos en cuanto al sueño, que hacen que se incremente la ansiedad y preocupación.
Pero estas terapias y los cambios en los estilos y hábitos de vida no son suficientemente eficaces en todos los pacientes ni en todos los tipos de insomnio, por lo que es común considerar la asociación de una segunda línea de tratamiento farmacológico (primera opción en los casos más graves), para lo cual se debe tener en cuenta tanto el tipo como la duración del trastorno y la influencia en la vida del sujeto.
Sin olvidar que la farmacoterapia de las patologías concomitantes no es excluyente sino complementaria en algunos casos, los fármacos hipnóticos actualmente disponibles frente al in-somnio tienen diferencias notables en su perfil beneficio-riesgo. Su uso se fundamenta en los mecanismos fisiopatológicos que subyacen al insomnio primario (no del todo conocidos), de modo que pretenden modificar en uno u otro sentido el desequilibrio entre la inhibición de los sistemas mantenedores de la vigilia y los encargados de activar los sistemas generadores del sueño. A grandes rasgos, la mayoría de los agentes farmacológicos ejercen un efecto potenciador del sistema inhibitorio gabaérgico (benzodiazepinas, los fármacos Z, extractos de valeriana), si bien otras estrategias buscan interferir con la regulación de los ciclos de sueño/vigilia (melatonina) o bien se aprovechan de la observación de la somnolencia inducida por algunos principios activos originariamente comercializados con otro fin (difenhidramina y doxilamina).
La selección individualizada de uno u otro medicamento estará condicionada por diversos factores, como la respuesta a tratamientos pasados, el objetivo terapéutico, el perfil de eficacia y tolerabilidad, los síntomas del paciente o sus comorbilidades, considerando para la selección óptima del tratamiento que el insomnio es un problema no solo nocturno, sino que afecta a la calidad de vida y funcionalidad diurna.
En líneas generales, el uso de agentes hipnóticos debe realizarse en monoterapia, a la menor dosis posible y durante periodos cortos o de forma intermitente. Pero, incluso con ese uso “ideal”, estos fármacos pueden empeorar el rendimiento diurno y provocar efectos adversos a corto y largo plazo. El empleo de antidepresivos o antipsicóticos sedativos está solo justificado cuando existe una patología concomitante en la cual esté indicado el empleo de estos medicamentos.
En muchos casos, los pacientes que consultan por insomnio crónico llevan haciendo un uso prolongado de depresores del SNC como las benzodiazepinas (BZD)4, hecho que hay que intentar evitar, ya que hay estudios que demuestran que en España se produce un abuso de prescripción y consumo de estos fármacos agonistas gabaérgicos no selectivos5, que conllevan un importante problema asociado de tolerancia y un importante potencial de abuso con su uso continuado. Las BZD más empleadas por vía oral en el abordaje del insomnio son las de duración de acción corta-media lorazepam y lormetazepam, que carecen de metabolitos activos y tienen una semivida plasmática de eliminación de 8-12 h. Con indicación y uso principal como hipnóticos también destacan: triazolam, una BZD de acción ultracorta de < 6 h y semivida de 2-4 h, y flurazepam, que representa una excepción, ya que, si bien la semivida del fármaco original es de solo 1 h, se considera una BZD de acción larga como consecuencia de la prolongada semivida de su metabolito desmetilado (60 h). En menor medida se emplean, en esa indicación autorizada, las siguientes BZD: brotizolam, clorazepóxido, clordiazepato potásico, loprazolam o quazepam. Diazepam, por su parte, no suele recomendarse para tratar el insomnio, pero es útil si se este trastorno se asocia con ansiedad diurna.
Tampoco están exentos del riesgo de dependencia física y/o psíquica ni del potencial de abuso los fármacos Z, un grupo de fármacos con propiedades hipnóticas/sedantes similares a las de las BZD (pero con estructuras diferentes) y prácticamente desprovistos de efectos ansiolíticos, cuya clasificación bajo este término proviene del hecho de que todos ellos comienzan por esta letra: zolpidem, zopiclona y zaleplón (no comercializado en España). Estos, que actúan también sobre el sitio receptor de las benzodiazepinas en varios subtipos del receptor GABAA y potencian los efectos inhibitorios del GABA, se indican únicamente en el tratamiento a corto plazo del insomnio transitorio y crónico en adultos (dificultades para iniciar el sueño, despertares nocturnos y despertar precoz), específicamente en aquellas situaciones graves o incapacitantes, como la asociación de ansiedad grave.
Los antagonistas de los receptores H1 de histama difenhidramina y doxilamina son fármacos de menor potencia hipnótica que las BZD, pero que, sin embargo, se usan ampliamente en España por incluirse en medicamentos sin receta médica. Sus efectos hipnóticos aparecen dentro de la primera hora tras la administración, por lo que suele recomendarse un comprimido aproximadamente media hora antes de acostarse, desaconsejándose su uso continuo durante más de 1 o 2 semanas. La evidencia disponible respalda que los anti-H1 pueden tener un papel en el tratamiento del insomnio ocasional a corto plazo en los adultos más jóvenes, pero también se asocian con una serie de inconvenientes: la tolerancia se desarrolla rápidamente, pueden causar una disminución del rendimiento laboral –por somnolencia residual– al día siguiente de su uso, y presentan un marcado efecto anticolinérgico, con riesgo de sequedad de boca, visión borrosa y estreñimiento.
Por otra parte, la administración exógena de melatonina (hormona implicada en la inducción del sueño y la regulación de los ritmos circadianos) ejerce un efecto hipnótico probablemente por la combinación de varios mecanismos: reducción de la temperatura corporal, modificación de los niveles cerebrales de neurotransmisores de tipo monoamina, normalización de los ritmos circadianos y efectos diversos sobre los receptores GABAA (potenciación en los localizados en el núcleo supraquiasmático e inhibición en los hipotalámicos). Fue inicialmente autorizada en comprimidos de liberación prolongada para el tratamiento a corto plazo –máximo de 13 semanas– del insomnio primario caracterizado por un sueño de mala calidad en adultos mayores de 55 años (edad a partir de la cual existe un descenso fisiológico de su producción y liberación), aunque muestra una eficacia modesta tanto sobre el tiempo de latencia del sueño como sobre su duración y calidad. Más recientemente se ha aprobado su uso en el tratamiento del insomnio en niños y adolescentes (población en la que el resto de opciones farmacológicas no tienen aprobación) con trastorno del espectro del autismo y/o síndrome de Smith-Magenis.
Tampoco se debe olvidar el cierto potencial terapéutico que frente al insomnio puede tener la fitoterapia, habiéndose descrito para distintas plantas medicinales, tales como valeriana, tila, melisa o pasiflora, entre otras (Fernández-Moriano, 2021).
Hasta ahora, según recogen las guías europeas de práctica clínica para la farmacoterapia del insomnio en adultos, únicamente se recomienda el uso de las BZD y otros hipnóticos no-benzodiacepínicos a corto plazo (< 4 semanas) y siempre y cuando haya afectación severa de la funcionalidad durante el día, pues, si bien favorecen la inducción al sueño, alteran su arquitectura y provocan en muchos casos tolerancia, dependencia e insomnio de rebote. En el manejo del insomnio crónico (> 4 semanas) se debe evitar el uso a largo plazo de fármacos hipnóticos y otros sedantes, de plantas medicinales o de otras terapias alternativas, y se recomienda optar en primera línea por intervenciones de tipo cognitivo conductual (TCC), acaso con un curso corto de fármacos hipnóticos para el manejo agudo de exacerbaciones del insomnio persistente; para pacientes de 55 años o más, se puede considerar una segunda línea con melatonina (NICE, 2015). Suele ser conveniente la derivación del paciente a una unidad clínica especializada en sueño si el insomnio persiste a pesar de las medidas no farmacológicas iniciales.
Acción y mecanismo
Daridorexant es un antagonista dual del receptor de la orexina, que actúa sobre los receptores orexina 1 y orexina 2 con igual potencia antagonista, de modo que bloquea el efecto de promoción de la vigilia que los neuropéptidos orexina A y orexina B ejercen al actuar sobre ellos. Así pues, por esa capacidad de daridorexant de evitar la activación de los receptores de la orexina, reduce el desencadenante de la vigilia (en vez de inducir el sueño mediante una amplia inhibición de la actividad cerebral, como otros fármacos hipnóticos) y facilita el sueño sin alterar la estructura del sueño, motivo por el cual el medicamento ha sido autorizado para el tratamiento por vía oral de pacientes adultos con insomnio caracterizado por presencia de síntomas durante al menos 3 meses e impacto considerable en la actividad diurna.
Se ha hipotetizado que daridorexant actúa tanto sobre el mantenimiento del sueño como sobre el tiempo de latencia de sueño, a diferencia de otros hipnóticos como las benzodiazepinas o los fármacos Z, que inciden fundamentalmente en el tiempo de latencia. La preservación con el nuevo fármaco de la estructura del sueño en todas sus fases ha sido confirmada mediante registros electroencefalográficos en roedores y mediante pruebas de polisomnografía en pacientes con insomnio. Además, gracias a su semivida optimizada carece de efectos residuales a la mañana siguiente.
Los estudios preclínicos en modelo de rata han permitido evidenciar la capacidad de daridorexant para atravesar la barrera hematoencefálica y acceder al cerebro a dosis farmacológicamente activas, que redujeron entre un 26% y un 74% el tiempo total de sueño. Tanto en ratas como en perros, el fármaco redujo de forma dosis-dependiente la vigilia y promovió el sueño (redujo la latencia hasta un sueño no-REM persistente y aumentó el tiempo de sueño tanto REM como no-REM en torno a un 39%, sin modificar sus proporciones en el tiempo de sueño total), con un inicio de la somnolencia en la primera hora tras la administración y un efecto que se prolongó durante al menos 6 h.
Una vez en nuestro organismo, daridorexant tiene 3 principales metabolitos que también actúan como antagonistas de los receptores de orexina 1 y 2, aunque con menor potencia (mayor valor de Kb) que el fármaco original. Los estudios de simulación sugieren un pico intenso y rápido de ocupación por el fármaco de los receptores de los receptores de orexinas y también una veloz bajada en la proporción de ocupación de dichos receptores (EMA, 2022).
Aspectos moleculares
El nombre químico del clorhidrato de daridorexant es (S)-2-(5-cloro-4-metil-1H-benzo[d]imidazol-2-il)-2-metilpirrolidin-1-ilmetanona, que se corresponde con la fórmula molecular C23H23ClN6O2.HCl y un peso molécula de 487,4 g/mol en su forma de sal (450,9 g/mol como base libre). El principio activo se presenta como un polvo cristalino no higroscópico de color blanco a amarillo claro, muy ligeramente soluble en medios ácidos pero insoluble a pH neutro.
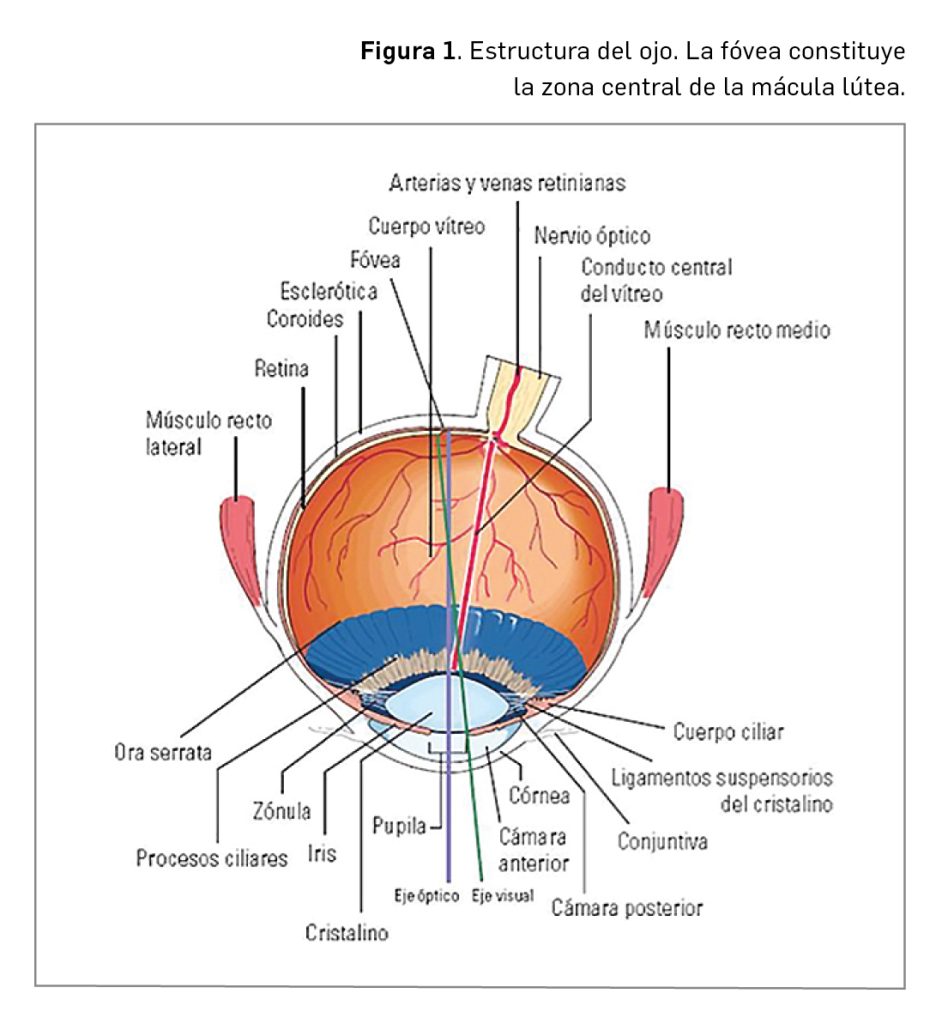
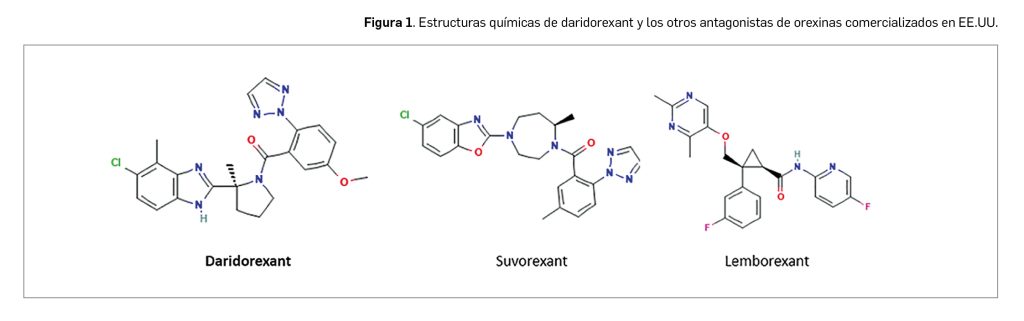
Su estructura química (Figura 1) contiene un único centro quiral. Es el único antagonista de las orexinas hasta ahora autorizado en Europa, si bien en EE.UU. hay otros dos autorizados (suvorexant y lemborexant), que tienen semividas plasmáticas superiores (de 8 h en el caso de daridorexant, frente a 17 h y 12 h, respectivamente). Daridorexant es el resultado de un programa de optimización molecular, y ha sido seleccionado en base a estudios de modelización farmacocinética y farmacodinámica a partir de 20 análogos estructurales escogidos de entre más de 25 000 moléculas sintetizadas.
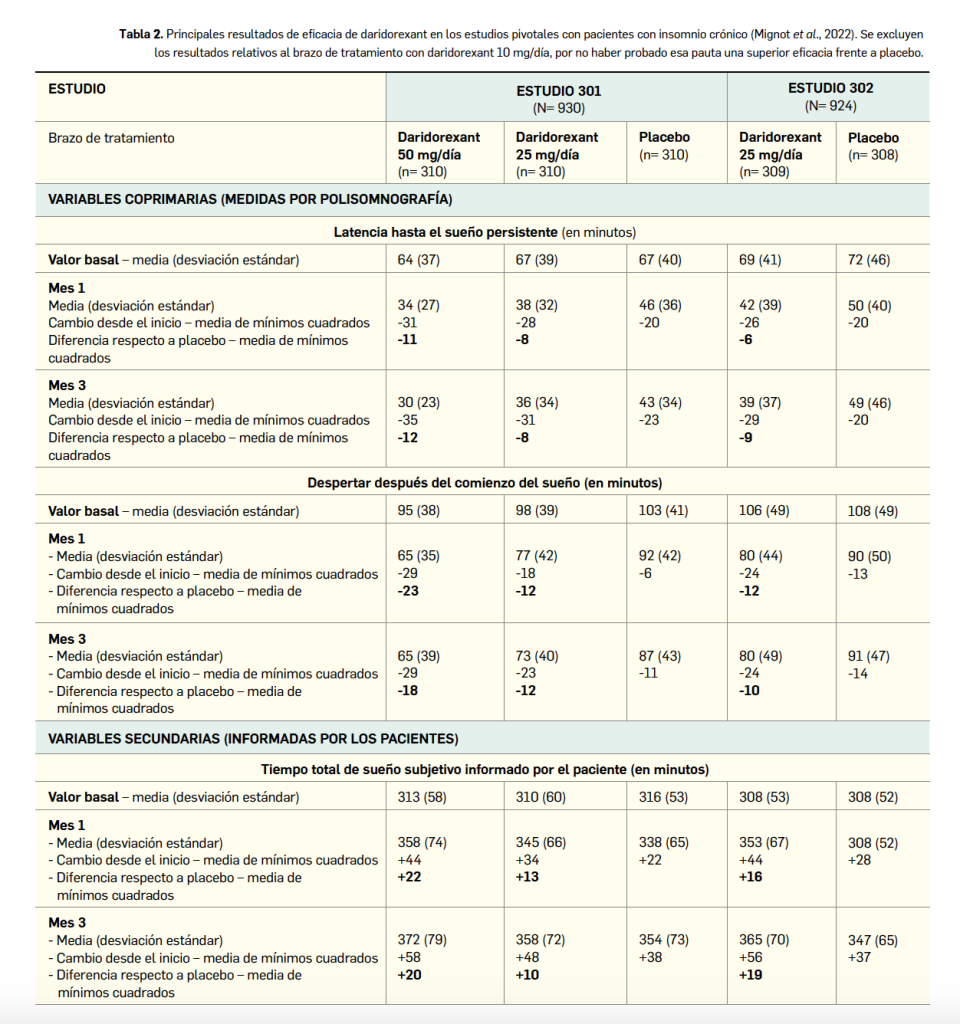
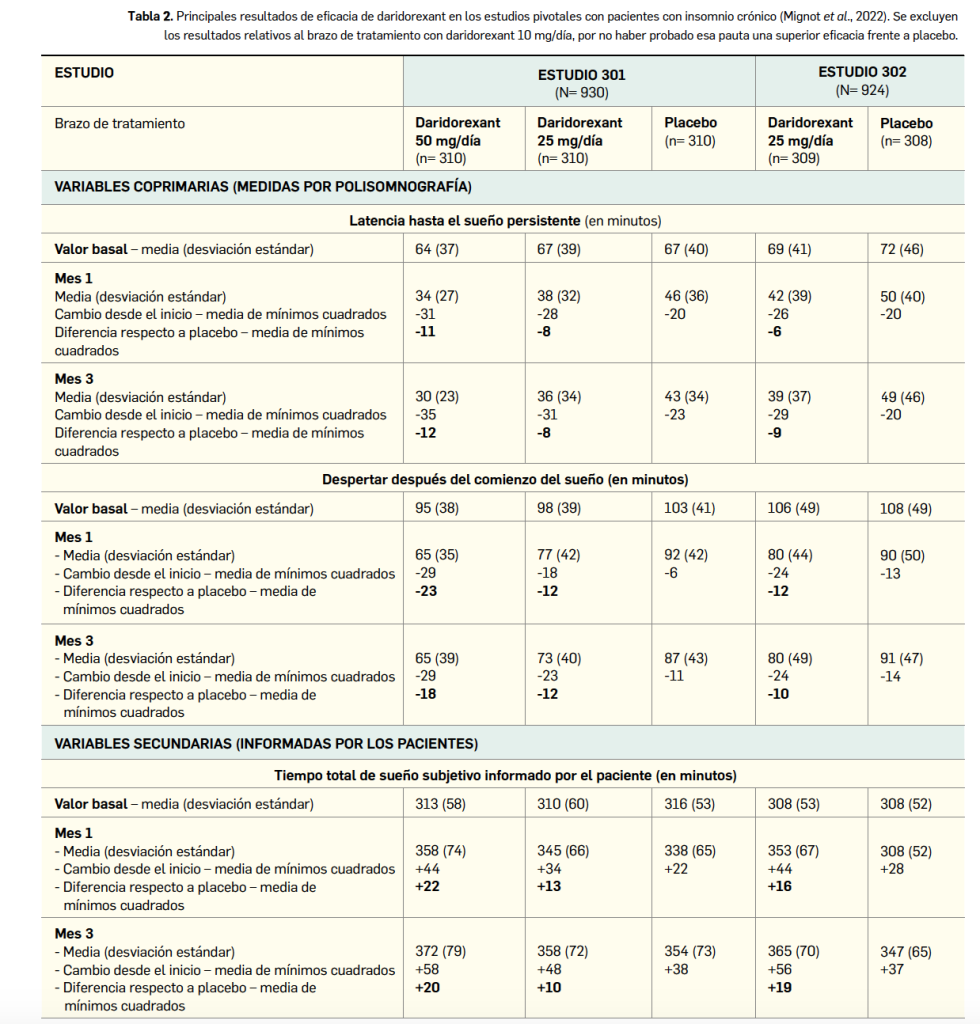
La eficacia y seguridad clínica de daridorexant han sido adecuadamente contrastadas en dos amplios ensayos pivotales confirmatorios de fase 3 (estudios 301 y 302) e idéntico diseño: doblemente ciegos, multicéntricos y multinacionales (156 centros en 17 países), aleatorizados, de grupos paralelos y controlados por placebo. En ellos se enrolaron un total de 1854 pacientes (930 y 924, respectivamente) con insomnio –definido como insatisfacción con la cantidad o calidad del sueño durante ≥ 3 meses y dificultad o deterioro clínicamente significativo de la actividad diurna–, quienes fueron asignados al azar en tres grupos de tratamiento a recibir daridorexant a la dosis de 25 mg/día, otra dosis (50 mg/día en el estudio 301 y 10 mg/día en el estudio 302) o un placebo equivalente, siempre administrados por la noche y durante 3 meses (84 días).
Considerados en conjunto, ya que las características demográficas y clínicas basales eran muy similares entres estudios y brazos de tratamiento, los pacientes presentaron una edad media de 56 años (intervalo de 18 a 88 años, un 39% tenía ≥ 65 años y un 6% > 75 años), una amplia mayoría eran mujeres (68%), y la proporción de sujetos con un índice de gravedad del insomnio6 (ISI) entre 8 y 14 puntos (insomnio subclínico), entre 15 y 21 puntos (insomnio moderado) y entre 22 y 28 puntos (insomnio grave) fue del 12%, 58% y 30%, respectivamente.
En ambos estudios se tomaron como variables coprimarias de eficacia el cambio respecto al inicio en la latencia hasta el sueño persistente (LSP) y en el despertar después del comienzo del sueño (WASO, por sus siglas en inglés) a los meses 1 y 3, objetivadas ambas por polisomnografía en un laboratorio de sueño. Como variables secundarias se investigaron al final del mes 1 el tiempo total de sueño subjetivo registrado por el paciente (TTS, medido cada mañana en casa mediante un cuestionario diario del sueño) y la actividad diurna informada por el paciente (medida cada noche mediante el dominio de la tendencia al sueño del cuestionario validado IDSIQ de síntomas diurnos e impacto del insomnio), entre otras.
Los principales resultados divulgados (Tabla 2; Mignot et al., 2022) demuestran que la eficacia de daridorexant es significativamente superior a placebo con la mayor dosis (p< 0,001), tanto sobre las variables de sueño objetivas (primarias) como sobre las subjetivas, y en los dos puntos temporales considerados (mes 1 y mes 3). La dosis de 10 mg/día del fármaco no fue eficaz. De modo interesante, esos hallazgos fueron consistentes en los subgrupos evaluables de pacientes, tanto para las variables primarias como para las secundarias, con independencia de factores como la edad, el sexo o la raza.
Tras el periodo de 3 meses de tratamiento en los estudios, los pacientes fueron sometidos a un lavado del fármaco de 7 días con placebo, durante el cual se investigó el potencial de insomnio de rebote: se vio que los valores medios en las variables de eficacia coprimarias eran mejores que los basales (-15 min en la latencia de sueño y -3 min en el despertar tras el inicio del sueño en los pacientes tratados con la dosis recomendada de 50 mg), lo cual indica que no hay signos de insomnio de rebote tras la suspensión de daridorexant.
Tras el lavado se ofreció a los pacientes participar en un estudio de extensión, también doble ciego y controlado por placebo, de 9 meses de duración (estudio 3). Así, un buen número de sujetos (n= 576) recibió el fármaco durante al menos 6 meses, incluyendo 331 pacientes que fueron tratados durante 1 año. Los resultados del estudio de extensión no se han divulgado aún, pero el EPAR de la EMA sugiere que la eficacia se mantiene en magnitud similar tras 1 año, de manera consistente en las distintas variables evaluadas (EMA, 2022).
Desde el punto de vista de la seguridad, los principales datos derivan también de los citados ensayos pivotales y el de extensión, en que casi 1900 pacientes con insomnio han recibido tratamiento con daridorexant; más de 300 lo han recibido durante al menos 1 año. La incidencia global de eventos adversos fue solo ligeramente mayor con el nuevo fármaco que con placebo (41-43% vs. 36-37%), si bien los considerados relacionados con el tratamiento que fueron graves o condujeron a discontinuación fueron algo más frecuentes con placebo (2,3-3,2%) que con daridorexant (1-2,3%, sin relación con la dosis), lo cual es reflejo de su perfil toxicológico relativamente benigno.
Las reacciones adversas más reportadas con el uso del fármaco (< 10%), con una incidencia comparable a la del grupo control, fueron cefalea (4-6% vs. 4% con placebo), somnolencia (2-3% vs. 2%) y nasofaringitis. La fatiga, las náuseas y los mareos (< 2-3%) tuvieron cierta mayor frecuencia numérica con daridorexant, pero sin diferencias notables. La práctica totalidad de ellas fueron leves-moderadas en severidad, considerándose graves solo un 1-2% de los eventos adversos. Además, cabe destacar que, durante el periodo inicial de 3 meses de tratamiento, solo se reportó una muerte como consecuencia de un evento adverso, que no se relacionó con el fármaco. Las alteraciones del sueño, como la parálisis o alucinaciones relacionadas con el sueño, tuvieron una muy baja incidencia (< 1%, sobre todo durante las primeras semanas), como también fue muy baja la tasa de discontinuación por eventos adversos entre los pacientes tratados con daridorexant (1% vs. 2,3% con placebo). De modo interesante, este perfil de seguridad parece mantenerse constante durante al menos 1 año y también es comparable entre grupos etarios (Kunz et al., 2023).
Aspectos innovadores
Daridorexant es un novedoso antagonista del receptor de la orexina, que ejerce una acción dual y equipotente sobre los receptores orexina 1 y orexina 2, bloqueando el efecto promotor de la vigilia que los neuropéptidos orexina A y orexina B ejercen al actuar sobre ellos. Se diferencia, pues, de otros fármacos hipnóticos en que no provoca una inhibición generalizada de la actividad cerebral. Por su capacidad de evitar la activación de los receptores de la orexina y reducir el desencadenante de la vigilia, facilita el sueño sin alterar su estructura normal, siendo esta la base de la autorización del medicamento para el tratamiento por vía oral de pacientes adultos con insomnio caracterizado por presencia de síntomas durante al menos 3 meses e impacto considerable en la actividad diurna.
Los datos conducentes a su aprobación han derivado de dos amplios estudios pivotales casi idénticos, considerados por la EMA de adecuado diseño (doble ciego, multicéntrico, aleatorizado y controlado por placebo). En ellos la pauta recomendada (50 mg/día) ha probado una eficacia significativamente superior frente a placebo en variables objetivas y subjetivas que reflejan convenientemente la severidad del insomnio y su impacto en la funcionalidad diurna.
Un tratamiento de un mes con el fármaco redujo significativamente el tiempo de latencia del sueño –que al inicio rondaba 1 h de media– entre 6 y hasta 12 min frente a placebo, con una magnitud del efecto constante tras 3 meses. Por ejemplo, tras 1 mes, en uno de los estudios se vio que los pacientes tratados con daridorexant conciliaban el sueño 35 min más rápido que antes del inicio (vs. 23 min más rápido en el brazo control). Si se asume como respuesta clínica el objetivo ambicioso de reducir 25 min la latencia, la tasa de respondedores con daridorexant es del 56% y del 45% con placebo. También en comparación con placebo, el tiempo despierto tras el inicio del sueño –que en el estado basal superaba la hora y media– se vio reducido en un promedio entre 12 y 23 min, tanto al mes 1 como al 3; a modo ilustrativo, se puede subrayar que, tras 1 mes de tratamiento, el fármaco redujo el tiempo que pasaban despiertos cada noche en 29 min (vs -11 min en los que recibieron placebo). Tomando la barrera de -30 min como una respuesta clínicamente relevante, se estima que responden un 50% de los pacientes tratados con daridorexant y un 28% con placebo. En ambas variables coprimarias, relativas a la inducción y al mantenimiento del sueño, respectivamente, la dosis diaria de 50 mg se muestra más eficaz que la de 25 mg, que se asocia a una eficacia cuestionable.
Por su parte, las variables secundarias respaldan el beneficio del fármaco. Se constató una mejoría de la duración total subjetiva del sueño, que rondaba al inicio las 5 h, de entre 10 y 22 minutos frente a placebo, mayor para la dosis alta y mantenido durante los 3 meses de evaluación. Habiéndose reportado una mejora de hasta +58 min al tercer mes con daridorexant, se vio una tasa de respuesta (entendida como incremento de +55 min) del 48% con la dosis alta del fármaco y del 37% con placebo. Además, el impacto sobre la actividad diaria de los pacientes mejora notablemente, según se refleja en la puntuación del dominio de tendencia al sueño del cuestionario IDSIQ, que disminuye de media 5 puntos tras 3 meses de tratamiento, superior en casi 2 puntos a placebo.
Entre las limitaciones de la evidencia disponible se puede citar que la dosis recomendada solo se investigó en uno de los estudios pivotales, si bien su eficacia parece bien contrastada. Asimismo, las guías clínicas de la EMA en insomnio recomiendan la inclusión de un comparador activo en al menos un estudio de fase 3, lo cual no se cumplió en el desarrollo de daridorexant. No obstante, el uso crónico planteado para el fármaco podría explicar el uso de placebo, ya que la adición de zolpidem como control activo solo durante el primer mes habría complicado los análisis comparativos y la interpretación de resultados (EMA, 2022).
Por lo que respecta a la seguridad, se trata de un fármaco con un perfil toxicológico aceptable y benigno, en el que predominan, con una frecuencia baja (en todos los casos < 6%) y semejante a placebo, reacciones adversas inespecíficas, tales como cefalea, somnolencia, nasofaringitis, fatiga, náuseas y mareos. Casi todas ellas son leves-moderadas y determinan una tasa muy baja de interrupciones por motivos de seguridad, acaso incluso más baja con daridorexant que con placebo (1% vs. 2%).
El periodo de lavado de 1 semana posterior a los 3 meses de duración de los estudios permitió descartar la aparición de insomnio de rebote y el riesgo de síndrome de abstinencia con el nuevo fármaco, indicativo de la ausencia del fenómeno de dependencia, que permitirá detener el tratamiento sin medidas específicas. Esto, unido a su buena tolerabilidad y al mantenimiento del perfil de seguridad, y también de la eficacia, en periodos prolongados de al menos 1 año, constituyen quizás las principales ventajas que incorpora frente a las alternativas disponibles en la farmacoterapia del insomnio (benzodiazepinas, fármacos Z, antihistamínicos, etc.), que fueron desarrollados para su uso durante menos de 1 mes e inciden casi exclusivamente sobre la latencia del sueño.
En definitiva, se trata de un innovador fármaco que inaugura una vía terapéutica en su indicación. Con un inicio rápido de su efecto farmacológico, reduce el exceso de alerta en la vigilia, habiéndose probado superior a placebo en la inducción y el mantenimiento del sueño, sin afectar a la estructura del mismo ni a la capacidad funcional de la persona al día siguiente. Constituye la primera incorporación al arsenal terapéutico del insomnio en más de dos décadas, y el primero que aborda de manera específica la fisiopatología y ha ganado indicación en insomnio crónico, por ser compatible con un uso a largo plazo (aunque lo ideal es la mínima duración del tratamiento, que debe reevaluarse cada 3 meses). Se prevé, por tanto, que pueda suponer una modificación importante de la terapéutica estándar en pacientes con insomnio crónico (entre el 8% y el 12% de la población) y de alto impacto en su actividad diurna. En cualquier caso, su eficacia parece moderada y hay que recordar que se posicionará como una segunda línea de tratamiento tras la intervenciones cognitivo-conductuales.