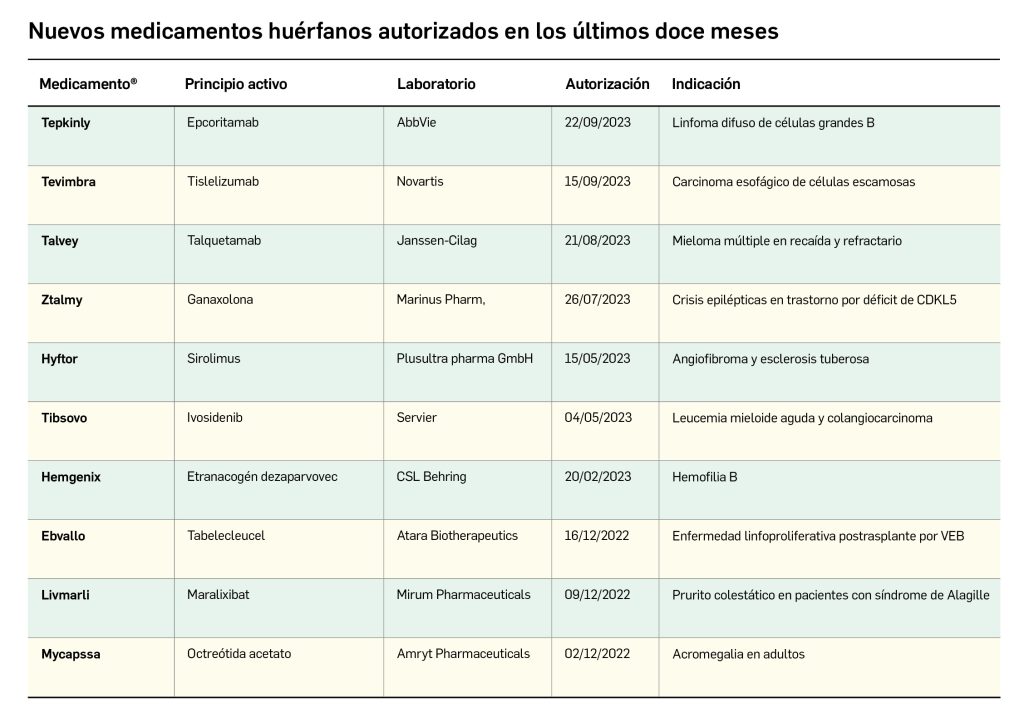
Los medicamentos huérfanos son aquéllos que sirven para diagnosticar, prevenir o tratar enfermedades raras de carácter muy grave o con riesgo para la vida. En la Unión Europea, la calificación de enfermedad rara se aplica a todas aquellas que no afectan a más de 5 de cada 10.000 personas. La designación de un medicamento como huérfano no garantiza su uso en la condición designada y no implica necesariamente que el producto satisfaga los criterios de eficacia, seguridad y calidad necesarios para la concesión de la autorización de comercialización. Como para cualquier medicamento, estos criterios solo pueden ser evaluados una vez que la solicitud de autorización de comercialización haya sido presentada.
Instituciones y redes españolas
Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación):
La autoridad reguladora de medicamentos de Nueva Zelanda (MedSafe) ha informado que se actualizará la información sobre neurotoxicidad en la información técnica de los medicamentos con cefalosporinas, en particular, casos de encefalopatía, mioclonía y/o convulsiones.
En Nueva Zelanda, la autoridad reguladora de medicamentos, MedSafe, ha anunciado que está trabajando con los titulares de registro sanitario de comercialización de medicamentos con cefalosporinas para actualizar las fichas técnicas, y así incluir datos sobre el riesgo de neurotoxicidad (MedSafe, 2023).
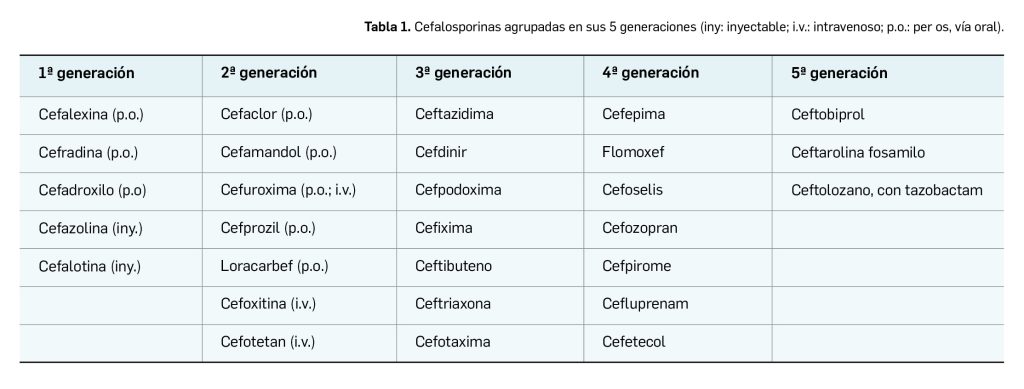
Las cefalosporinas son antibióticos betalactámicos de amplio espectro que se utilizan en atención primaria y secundaria para tratar una variedad de infecciones. Las cefalosporinas se agrupan en cinco generaciones según sus propiedades antibacterianas y su descubrimiento, si bien hay un cierto desacuerdo sobre la definición de generaciones (Werth, 2022). En la Tabla 1 se describen algunas de estas cefalosporinas.
Las cefalosporinas se ordenan en grupos llamados “generaciones” por sus características antimicrobianas y cronología de uso. Las primeras cefalosporinas fueron agrupadas en la “primera generación”, mientras que, más adelante, las cefalosporinas de espectro amplio fueron clasificadas como cefalosporinas de “segunda generación”. Cada nueva generación de cefalosporinas tiene más potencia frente a bacterias Gram-negativas, características antimicrobianas perceptiblemente mayores que la generación precedente; actualmente se diferencian cinco generaciones de cefalosporinas.
Cabe destacar que las cefalosporinas de primera generación tienen mayor espectro de acción contra estafilococo y estreptococo que las generaciones más recientes (Werth BJ, 2022).
Así, las cefalosporinas de 1ª generación tienen actividad predominante contra cocos grampositivos —Streptococcus y Staphylococcus—. Las cefalosporinas usuales no son activas contra cepas de Staphylococcus aureus resistentes a meticilina (SARM). Las de 2ª generación amplían el espectro incluyendo anaerobios y microorganismos Gram-negativos. Las bacterias del género Klebsiella suelen ser sensibles. Están indicadas en el tratamiento de la sinusitis, otitis, infecciones respiratorias bajas, peritonitis y diverticulitis. Debe evitarse su uso en infecciones por Enterobacter, y también la administración intramuscular debido al dolor. Todas ellas se eliminan por vía renal.
Las de 3ª generación suelen resultar más eficaces frente a los bacilos Gram-negativos y frente a los cocos Gram positivos (excepto S. aureus), que los fármacos de 1ª y 2ª generaciones. Son el tratamiento de elección en la meningitis por bacilos Gram-negativos y se utilizan también para combatir otras infecciones por bacilos Gram-negativos. Todo este grupo de 3ª generación son extremadamente activas contra la mayoría de las bacterias Gram-negativas (excepto Enterobacter y Citrobacter) incluyendo las mencionadas anteriormente, y contra bacterias productoras de beta-lactamasas. La ceftazidima y la cefoperazona son activas contra Pseudomonas aeruginosa, pero son menos activas que otros agentes de tercera generación contra cocos Gram-positivos. Estos antibióticos no están indicados en la profilaxis quirúrgica de rutina. Al igual que en los grupos anteriores hay parenterales y orales.
Las cefalosporinas de 4ª generación tienen un mayor espectro contra organismos Gram-positivos que las cefalosporinas de la 3ª generación. También tienen una mayor resistencia a beta-lactamasas que las cefalosporinas de la 3ª generación (Werth BJ, 2022).
Por último, las cefalosporinas de 5ª generación –como ceftobiprol (▼Zevtera®)– tienen mayor actividad contra Staphylococcus aureus resistente a meticilina (SARM), Streptococcus pneumoniae resistente a penicilinas, Pseudomonas aeruginosa y enterococos. La ceftarolina (Zinforo®) es activa tanto frente a Staphylococcus aureus como frente a estafilococos coagulasa negativos sensibles y resistentes a meticilina (SARM), así como frente a las recientes cepas resistentes a vancomicina (VRSA) y a daptomicina. También incluye en su espectro antimicrobiano a Streptococcus pneumoniae (incluyendo las cepas resistentes a penicilina), Haemophilus influenzae (incluyendo las cepas productoras de betalactamasas), Moraxella catarrhalis, Enterococcus faecalis, incluyendo cepas vancomicina-resistentes e inactivo frente Enterococcus faecium. Y, por último, ceftolozano (Zerbaxa®) se utiliza combinado con tazobactam, un inhibidor de las beta-lactamasas (Werth, 2022). Todas las cefalosporinas de 5ª generación han sido calificadas como de uso hospitalario en España.
En Nueva Zelanda, las notificaciones de casos y las revisiones de series de casos constataron que, en comparación con otras cefalosporinas, la cefepima se asoció con la mayoría de los informes de neurotoxicidad a nivel internacional (MedSafe, 2023). Sin embargo, se ha informado neurotoxicidad con todas las generaciones de cefalosporinas: cefazolina ha estado implicada en 7 casos, cefuroxima en 6 casos, cefaclor en 3, y cefalexina, cefotaxima, ceftazidima, ceftriaxona y cefepima en 2 casos respectivamente en cada una de ellas. Las notificaciones de neurotoxicidad con cefalosporinas se caracterizan principalmente por encefalopatía, mioclonías y/o convulsiones. Los factores de riesgo descritos incluyen: grupos de mayor edad, insuficiencia renal, trastornos subyacentes del sistema nervioso central y la administración por vía intravenosa.
El Comité de Reacciones Adversas a los Medicamentos de Nueva Zelanda (MARC, Medicines Adverse Reaction Committee) recomendó que los profesionales de la salud consideren la neurotoxicidad inducida por cefalosporinas en pacientes con los factores de riesgo mencionados anteriormente y una afección neurológica de nueva aparición e inexplicable. En tales casos, puede ser conveniente retirar el medicamento (MedSafe, 2023).
Recomendaciones
Se sabe que la excreción de las cefalosporinas es principalmente por el riñón, y el riesgo de reacciones tóxicas a estos medicamentos puede ser mayor en los pacientes con insuficiencia renal. Debido a que los pacientes de edad avanzada son más susceptibles a tener una función renal disminuida, se debe tener precaución en la selección de la dosis y se debe controlar la función renal (véase la sección 5.2. Propiedades farmacocinéticas de sus fichas técnicas). En pacientes de edad avanzada con insuficiencia renal a los que se les administró la dosis habitual de cefalosporina, se produjeron efectos adversos graves (véase sección 4.8. Reacciones adversas de sus fichas técnicas) que incluían encefalopatía reversible (trastornos de la conciencia, confusión, alucinaciones, estupor y coma), mioclonía, convulsiones (incluido el estado epiléptico no convulsivo) y/o insuficiencia renal.
En las fichas técnicas españolas de algunas de las cefalosporinas de uso por vía oral y por vía inyectable ya se describe (en la sección 4.4.) advertencias y precauciones especiales sobre este riesgo que han de tenerse en cuenta cuando se usen (AEMPS, 2023):
Insuficiencia renal. En la experiencia post-comercialización, se han descrito los siguientes efectos adversos graves: encefalopatía reversible (trastornos de la consciencia que incluyen confusión, alucinaciones, estupor y coma), mioclonías, crisis epilépticas (incluyendo estados epilépticos no convulsivos) y/o fallo renal. La mayoría de los casos se dieron en pacientes con insuficiencia renal que recibieron dosis de cefalosporina superiores a las recomendadas. En general, los síntomas de neurotoxicidad se resolvieron después de interrumpir la cefalosporina y/o después de la hemodiálisis; no obstante, algunos casos tuvieron un resultado fatal.
Estas consideraciones deben hacerse extensivas a todas las cefalosporinas actuales, aunque en algún caso los textos de sus fichas técnicas no estén actualizados. Para comprobar la fecha de la revisión del contenido del texto, se puede consultar el apartado “fecha de revisión del texto” al final de la ficha técnica. Estos documentos oficiales, como también lo son los prospectos, se pueden consultar para todas las marcas comerciales de los medicamentos con cefalosporinas actualmente comercializados en el Centro de Información Online de Medicamentos de la AEMPS (CIMA; disponible en: https://cima.aemps.es/cima/publico/home.html).
La Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) ha acordado en el seno del PRAC informar a los médicos prescriptores de los inhibidores de la kinasa Janus (JAK) abrocitinib (▼Cibinqo®), baricitinib (▼Olumiant®), filgotinib (Jyseleca®), tofacitinib (Xeljanz®) y upadacitinib (▼Rinvoq®) con recomendaciones actualizadas para minimizar los riesgos de neoplasias malignas, acontecimientos adversos cardiovasculares mayores, infecciones graves, tromboembolismo venoso y mortalidad con su uso.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha remitido a los médicos prescriptores de estos medicamentos, una comunicación directa (DHPC, por sus siglas en inglés de Direct Healthcare Professional Communication) a través del envío por parte de los laboratorios titulares de estos medicamentos, con las recomendaciones decididas y acordadas en el Comité Europeo de Farmacovigilancia (PRAC, por sus siglas del inglés Pharmacovigilance Risk Assessment Committee), que integran expertos de las 27 agencias nacionales de la Unión Europea.
Los inhibidores de JAK abrocitnib (▼Cibinqo®), filgotinib (Jyseleca®), baricitinib (▼Olumiant®), upadacitinib (▼Rinvoq®) y tofacitinib (Xeljanz®) están indicados para el tratamiento de diferentes enfermedades inflamatorias crónicas, tales como artritis reumatoide (AR), artritis psoriásica, artritis idiopática juvenil, espondilitis anquilosante, espondilo-artritis axial no radiográfica, colitis ulcerosa, dermatitis atópica y alopecia areata. Las indicaciones autorizadas varían en cada medicamento, tal y como reflejan sus respectivas fichas técnicas.
En marzo de 2021 se envió una carta de seguridad dirigida a los profesionales sanitarios o DHPC sobre Xeljanz® (tofacitinib) (AEMPS, 2021a), informando de que los resultados de un ensayo clínico finalizado (A3921133) realizado en pacientes con AR de 50 años o mayores, con al menos un factor de riesgo cardiovascular adicional, sugerían un mayor riesgo de acontecimientos adversos cardiovasculares mayores (MACE) y de neoplasias malignas (excluyendo cáncer de piel no melanoma) en pacientes tratados con tofacitinib en comparación con un inhibidor de TNF-alfa (Ytterberg et al., 2022).
Posteriormente, en julio de 2021 se envió una DHPC adicional para informar sobre una mayor incidencia de infarto de miocardio, cáncer de pulmón y linfoma con tofacitinib (Xeljanz®) en comparación con los inhibidores de TNF-alfa, observadas en el mismo ensayo clínico, así como sobre las recomendaciones adoptadas en la ficha técnica de tofacitinib (AEMPS, 2021b).
Los resultados preliminares de un estudio observacional (B023) realizado con otro inhibidor de JAK, baricitinib, también sugieren un mayor riesgo de MACE y tromboembolismo venoso (TEV) en pacientes con AR tratados con baricitnib en comparación con los tratados con inhibidores de TNF‑alfa.
En la Unión Europea, tras la finalización del procedimiento de revisión por parte de la EMA de los datos disponibles sobre los cinco inhibidores de JAK, se han adoptado las recomendaciones indicadas en la carta1, que se exponen a continuación. En consecuencia, serán actualizados la ficha técnica, prospecto y los materiales sobre prevención de riesgos para profesionales sanitarios y pacientes de estos medicamentos.
Posteriormente, la agencia británica (MHRA, Medicines and Healthcare products Regulatory Agency) también ha revisado esta situación y se están realizando cambios en la información de las fichas técnicas (SmPC; Summary of Product Characteristics) para todos los medicamentos inhibidores de JAK autorizados en el Reino Unido frente a enfermedades inflamatorias, para tener en cuenta la caracterización de riesgos actualizada y las medidas ampliadas de minimización de riesgos. La MHRA ha enviado también una DHPC para informar sobre estos cambios (MHRA, 2023).
Recomendaciones
Las recomendaciones se basan en la observación de una mayor incidencia de neoplasias malignas, acontecimientos adversos cardiovasculares mayores (MACE), infecciones graves, tromboembolismo venoso (TEV) y mortalidad en pacientes con artritis reumatoide (AR) y determinados factores de riesgo tratados con inhibidores de JAK, en comparación con los tratados con inhibidores del TNF-alfa.
Estos riesgos se consideran efectos de clase y son aplicables a todas las indicaciones inflamatorias y dermatológicas de los inhibidores de JAK.
Estos inhibidores de JAK solo deben utilizarse si no se dispone de alternativas terapéuticas adecuadas en pacientes:
de 65 años y mayores;
fumadores o exfumadores que fumaron durante un largo periodo de tiempo;
con otros factores de riesgo cardiovascular o para el desarrollo de neoplasias malignas.
Los inhibidores de JAK deben utilizarse con precaución en pacientes con factores de riesgo de TEV distintos de los mencionados anteriormente.
Se han revisado las recomendaciones posológicas para algunos grupos de pacientes con factores de riesgo.
Se recomienda realizar un examen dermatológico periódico a todos los pacientes.
Los profesionales sanitarios deben explicar a sus pacientes los riesgos asociados al uso de inhibidores de JAK.
El PRAC (Pharmacovigilance Risk Assessment Committee) es el Comité europeo para la Evaluación de Riesgos en Farmacovigilancia. En sus reuniones mensuales se deciden cambios en la información autorizada de las fichas técnicas y de los prospectos de los medicamentos europeos por motivos de seguridad.
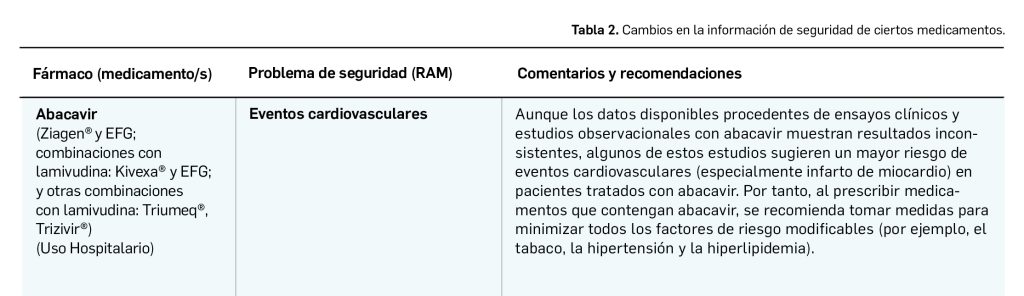
Una vez que se revisan y evalúan los datos de los informes periódicos de seguridad (IPS; en inglés PSUR), de forma colaboradora entre todas las 27 agencias nacionales, el Estado miembro principal que realiza la evaluación única de los IPS, o PSUSA (Periodic Safety Update report Single Assessment), propone los cambios y se aprueban en las reuniones mensuales del PRAC. Cuando afectan a medicamentos de registros nacionales se validan por el CMDh (Coordination Group for Mutual Recognition and Decentralised Procedures – Human), de la red de jefes de todas las Agencias de Medicamentos (HMA, Heads of Medicines Agencies) en sus reuniones mensuales, durante 3 días. Un procedimiento único, complejo y colaborativo de las 27 agencias nacionales de medicamentos de la Unión Europea. En la tabla siguiente se muestran los últimos cambios de información de seguridad acordados recientemente en el PRAC.
El Comité europeo para la Evaluación de Riesgos en Farmacovigilancia (PRAC) ha acordado cambios en las fichas técnicas y los prospectos de los siguientes medicamentos, siendo los más importantes los que se describen en la Tabla 2, según informa la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) en su Boletín mensual de Seguridad de Medicamentos de Uso humano de los meses de julio, agosto y septiembre de 2023 (AEMPS, 2023).
Las fichas técnicas y prospectos de los medicamentos pueden consultarse en la web de la AEMPS, dentro de la sección CIMA: Centro de Información Online de Medicamentos (https://cima.aemps.es/cima/).
Esta información que se incorpora a las fichas técnicas y prospectos de los medicamentos supone una actualización permanente, por lo que es necesario consultar sus datos y la fecha de la actualización (que figura al final del texto de las fichas técnicas y prospectos), cuando se consultan en la web de la AEMPS (sección CIMA: Centro de Información Online de Medicamentos).
Información importante
El Sistema Español de Farmacovigilancia de Medicamentos de Uso Humano (SEFV-H) se basa en el programa de notificación espontánea de un profesional sanitario (médico, odontólogo, farmacéutico, enfermero, otros) o de un ciudadano, de una sospecha de relación entre un medicamento (incluidos vacunas, sueros, gases medicinales, fórmulas magistrales, plantas medicinales) y un síntoma o signo adverso (reacción adversa, RAM) que manifieste el paciente o familiar (programa de tarjeta amarilla). El Real Decreto 577/2013 de Farmacovigilancia de medicamentos de uso humano (BOE núm. 179, de 27 de julio de 2013) entró en vigor el 28 de julio de 2013. La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) coordina el SEFV-H. A su vez se integra en el Sistema Europeo de Farmacovigilancia que desde 1995 coordina la Agencia Europea de Medicamentos (EMA), y participa desde 1984 en el Programa Internacional de Farmacovigilancia de la OMS, junto con más de 152 países.
¿Qué notificar? Se deben notificar las sospechas de RAM:
Con medicamentos autorizados, incluidas las de aquellos que se hayan utilizado en condiciones diferentes a las autorizadas o con medicamentos extranjeros importados con autorización de la AEMPS.
Principalmente las RAM ‘graves’ (mortales, o que amenacen la vida, prolonguen o provoquen una hospitalización, causen incapacidad o sean médicamente importantes y las trasmisiones de un agente infeccioso a través de un medicamento) o RAM ‘inesperadas’ de cualquier medicamento
Con medicamentos de ‘seguimiento adicional’ (durante sus primeros 5 años desde la autorización, identificados con un triángulo negro invertido (▼) a la izquierda del nombre del medicamento en el material informativo, en el prospecto y en la ficha técnica); ver la lista mensual de los medicamentos con “triángulo negro” en la web de la AEMPS: https://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/seguimiento_adicional.htm#lista_europea
Las que sean consecuencia de ‘errores de medicación’, que ocasionen daño en el paciente;
Las originadas por ‘interacciones’ con medicamentos, plantas medicinales, incluso alimentos (zumo de pomelo, ahumados, crucíferas, etc).
¿Cómo notificar?
No olvide notificar cualquier sospecha de RAM a su Centro Autonómico o Regional de Farmacovigilancia mediante las ‘tarjetas amarillas’. Consulte en este directorio su Centro Autonómico de Farmacovigilancia correspondiente.
MÉTODO electrónico: desde el 15 de enero de 2013 se puede notificar a través del sitio web https://www.notificaRAM.es/, y el sistema electrónico hace llegar a su centro correspondiente la notificación de sospecha de RAM. Sirve para profesionales sanitarios y para ciudadanos, en formularios diferentes. La nueva legislación europea de farmacovigilancia establece esta posibilidad para facilitar la notificación de las sospechas de RAM por la población en general.
¿Dónde conseguir tarjetas amarillas?
En Consultando a su Centro correspondiente del SEFV-H. Podrá encontrar el directorio de Centros en las primeras páginas del “Catálogo de Medicamentos” y en las páginas de Internet http://www.farmaceuticos.com y https://www.aemps.gob.es/medicamentos-de-uso-humano/directorio-de-centros-autonomicos-del-sistema-espanol-de-medicamentos-de-uso-humano-sefv-h/.
¿Dónde consultar las fichas técnicas y prospectos de los medicamentos?
En la página web de la AEMPS http://www.aemps.gob.es, seleccionando ”CIMA: Centro de Información on-line de Medicamentos de la AEMPS, Humanos”, se pueden consultar por nombre comercial o por sus principios activos. También están disponibles en la base de datos BOT PLUS.
NOTA: la mención de marcas comerciales en el texto solo tiene fines de identificación, y en absoluto se les debe asignar directamente lo descrito en el texto.
El Servicio de Dispensación está definido por Foro de Atención Farmacéutica en Farmacia Comunitaria (FORO AF-FC) como “el SPFA encaminado a garantizar que los destinatarios de los medicamentos y productos sanitarios, tras una evaluación individual, reciban y utilicen los medicamentos de forma adecuada a sus necesidades clínicas, en las dosis precisas según sus requerimientos individuales, durante el periodo de tiempo adecuado, con la información para su correcto proceso de uso y de acuerdo con la normativa vigente.”1
El farmacéutico, por tanto, ha de recabar información durante una entrevista sobre el destinatario de los medicamentos, con y sin receta, verificar que el medicamento es adecuado para ese destinatario (persona o animal) y no existen incidencias que no aconsejen la dispensación, así como comprobar que dispone de la información necesaria para una utilización efectiva y segura.
La información podrá obtenerse a través de la receta médica (en caso de medicamentos de prescripción), del destinatario del medicamento o cuidador, del sistema sanitario con la autorización del paciente o a través de los registros de la farmacia.
Disponer de una herramienta tecnológica en la que apoyarnos durante la dispensación (Figura 1) nos ayudará a realizar el procedimiento de una forma más sencilla, a identificar las incidencias con la medicación gracias a las alertas instantáneas que nos ofrece y a evidenciar nuestra labor profesional en torno al mismo, ya que de no utilizar una herramienta de registro no podremos dejar constancia de las intervenciones que realizamos desde la farmacia comunitaria, como por ejemplo, derivar al médico en el caso de haber detectado algún PRM/RNM o de una no dispensación de un medicamento, por diferentes motivos.
¿Qué objetivos tiene este servicio?
Garantizar el acceso a los medicamentos y entregarlos en las condiciones óptimas, de acuerdo con la normativa legal vigente.
Garantizar que el paciente o su cuidador conocen el proceso de uso de los medicamentos y que lo van a asumir.
Proteger al paciente frente a la aparición de RNM mediante la identificación y resolución de PRM.
Derivar a otros SPFA o a otros profesionales sanitarios, cuando proceda.
Mejorar la calidad de vida de los pacientes.
¿CÓMO PRESTAR ESTE SERVICIO A TRAVÉS DE NODOFARMA ASISTENCIAL?
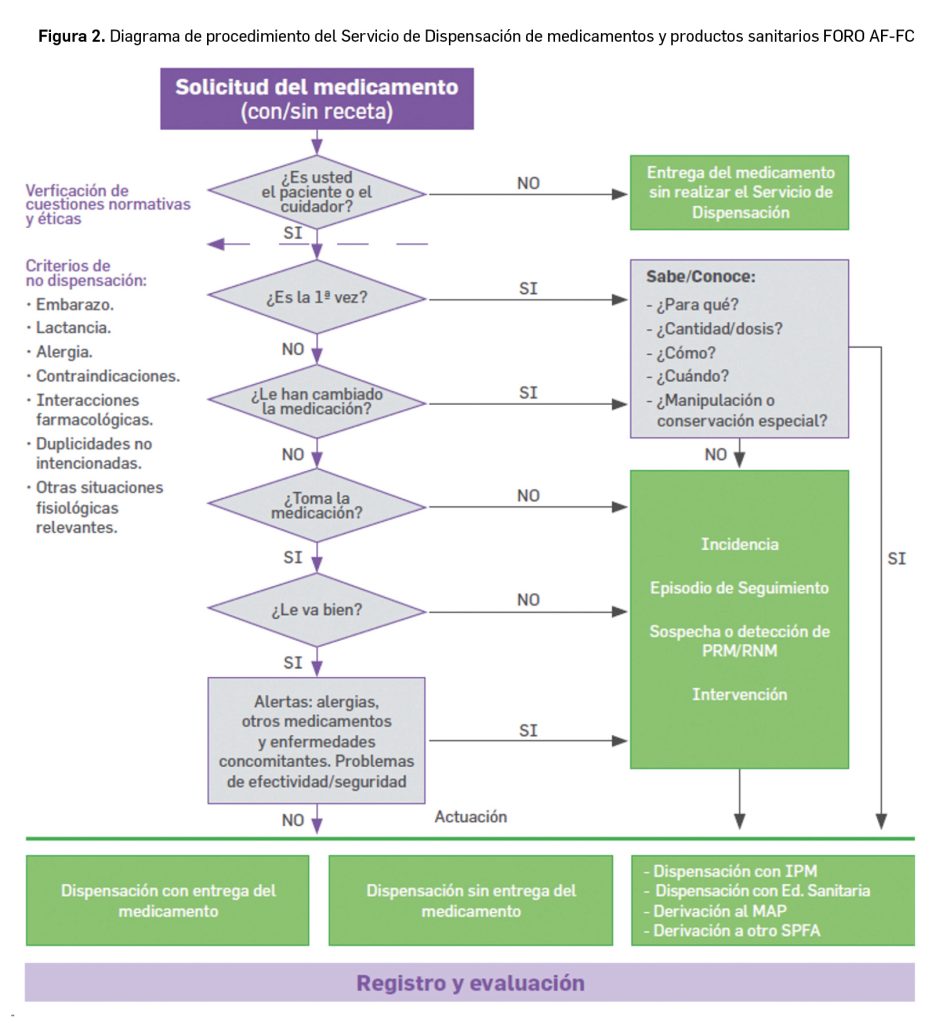
Para prestar este Servicio se sigue el procedimiento del Servicio propuesto por FORO AF-FC según la Figura 2.
A modo de resumen se siguen estos cuatro pasos:
Obtención de información sobre el paciente/cuidador y su farmacoterapia.
Evaluación de la información.
Actuación o intervención, en caso de Incidencia.
Registro y evaluación del proceso del Servicio.
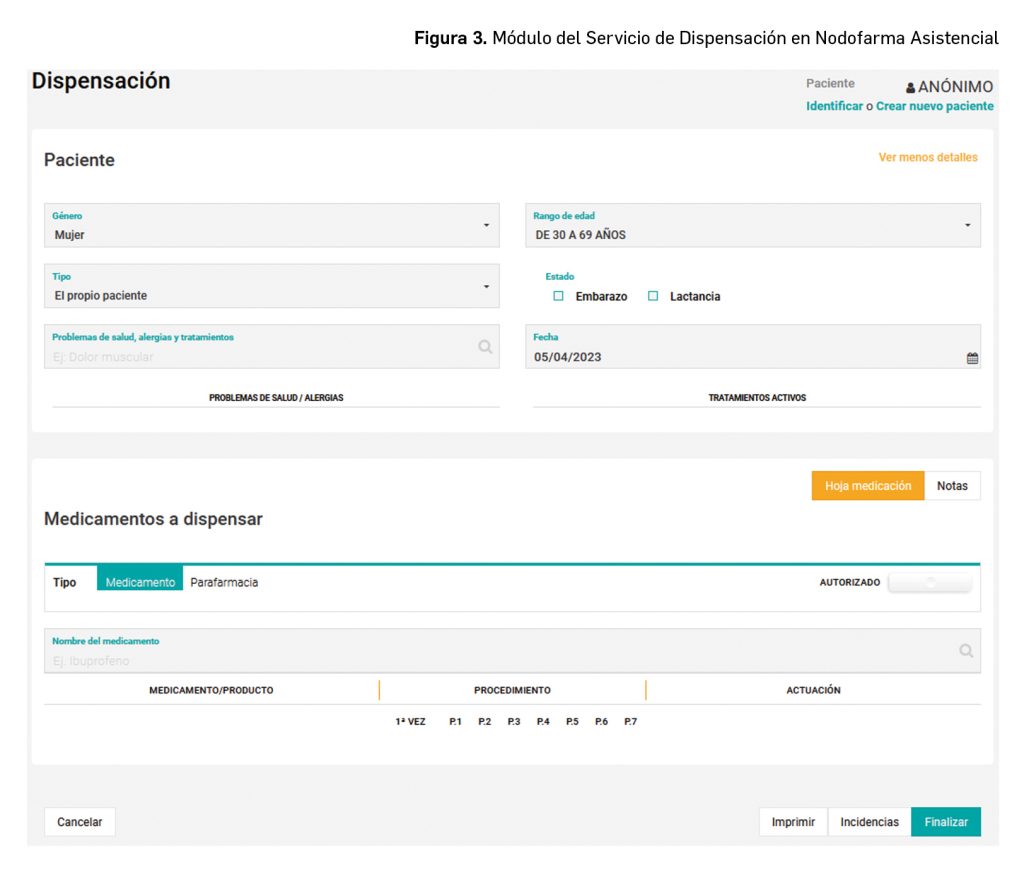
Al acceder al módulo Nodofarma Asistencial (Figura 3), permite identificar si el medicamento lo solicita el paciente, su cuidador, o una tercera persona considerando el sexo, la edad real/aproximada, y la relación con la persona que solicita el medicamento, además de si el paciente utiliza otros medicamentos de prescripción y/o automedicación, presenta enfermedades concomitantes y/o alergias, así como su situación fisiológica (embarazo/lactancia si aplica) que puedan afectar al tratamiento.
A continuación, la sección “Medicamentos a dispensar”, permite introducir los datos del medicamento o producto de parafarmacia a dispensar.
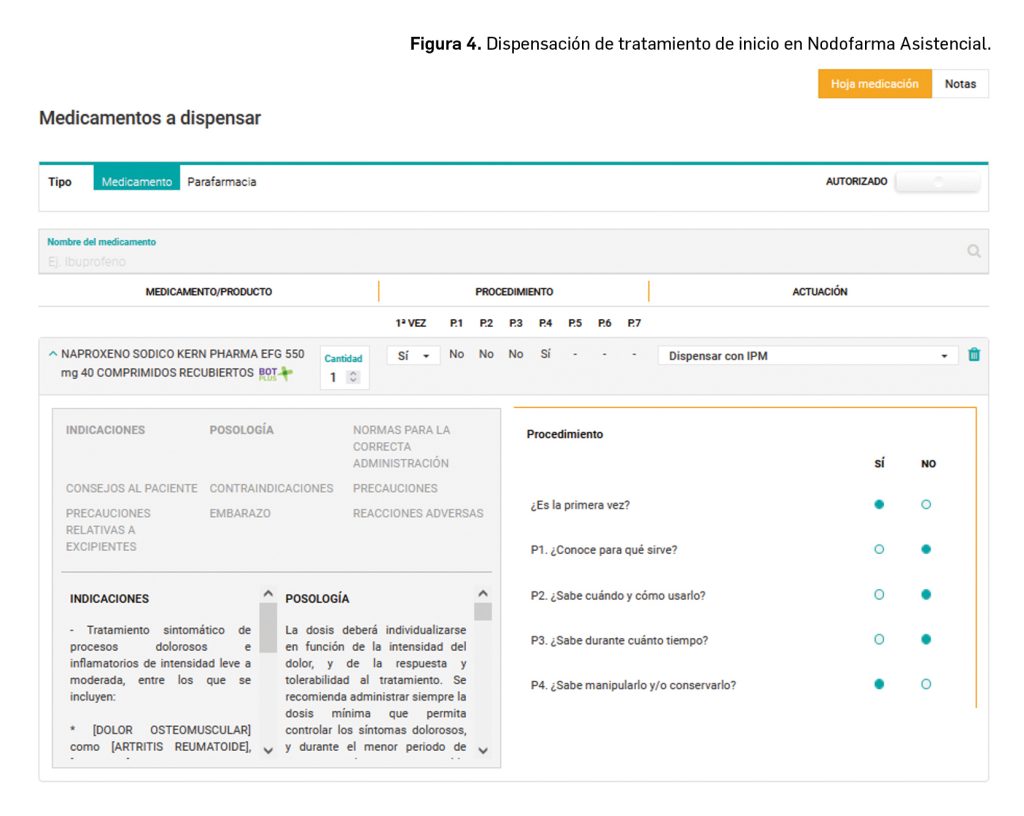
Si es la primera vez que la persona va a utilizar el medicamento habrá que realizar las siguientes preguntas con el objetivo de verificar y evaluar si el paciente o su cuidador conocen el correcto proceso de uso del medicamento (Figura 4):
Para qué lo va a usar.
Qué cantidad o dosis va a utilizar.
Cómo lo tiene que usar.
Durante cuánto tiempo lo va a usar.
Si sabe manipularlo y/o conservarlo (incluyendo la eliminación).
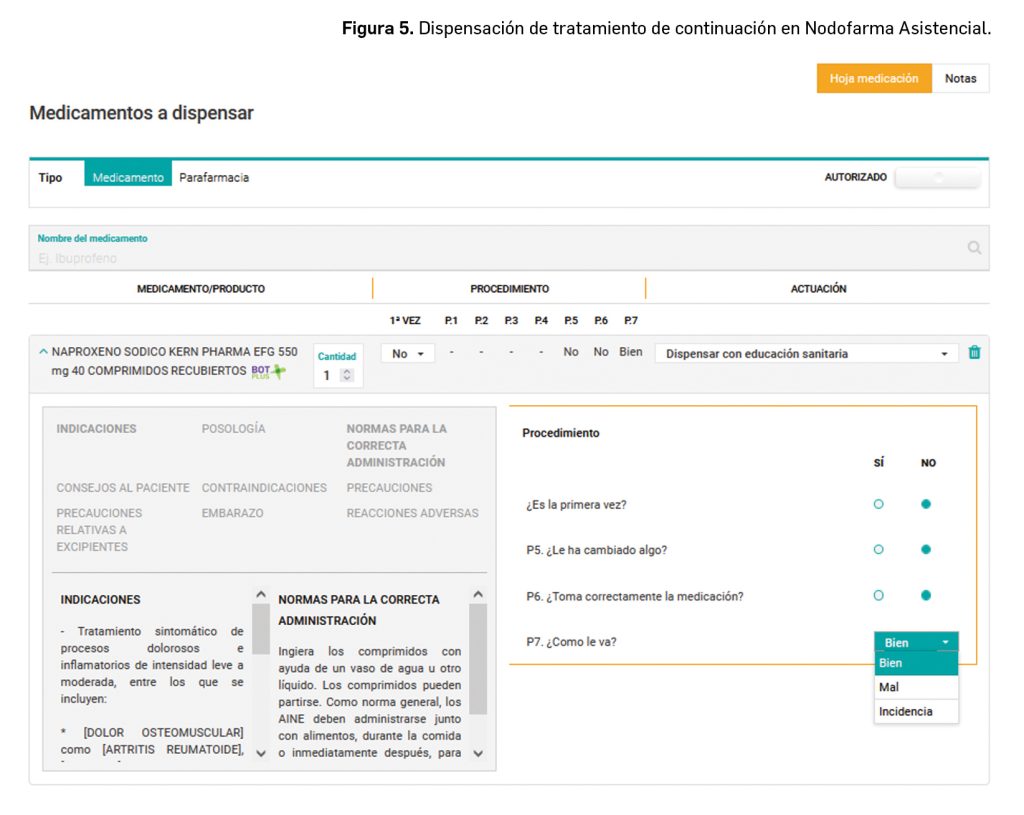
Si no es la primera vez que la persona va a utilizar el medicamento habrá que realizar las siguientes preguntas con el objetivo de valorar la percepción del paciente o su cuidador sobre la efectividad y seguridad del medicamento, haciendo un especial hincapié en la adherencia al tratamiento (Figura 5):
¿Está tomando la medicación como su médico le ha prescrito?
¿Le han cambiado algo? (pauta, dosis, etc.). *Si la respuesta es afirmativa, realizará las mismas preguntas que si se tratara de inicio de tratamiento.
¿Cómo le va el tratamiento?
Y/o ¿tiene algún problema con el tratamiento?
Con toda la información obtenida se podrán dar situaciones no esperadas que interrumpen el curso del Servicio, es decir, incidencias. En este caso, se identificarán Problemas Relacionados con los Medicamentos y se podrá identificar si el paciente sufre, o está en riesgo de sufrir, un problema de salud como consecuencia del uso o desuso de los medicamentos, es decir, un Resultado Negativo de la Medicación. Con ello se intervendrá en función de la situación:
Facilitar IPM.
Ofrecer educación sanitaria.
Derivar a otro SPFA.
Derivar al Médico de Atención Primaria (MAP) comunicando el PRM/RNM.
Derivar al MAP proponiendo cambios en el tratamiento.
Proponer otras modificaciones.
Notificar a farmacovigilancia de acuerdo a la legislación vigente.
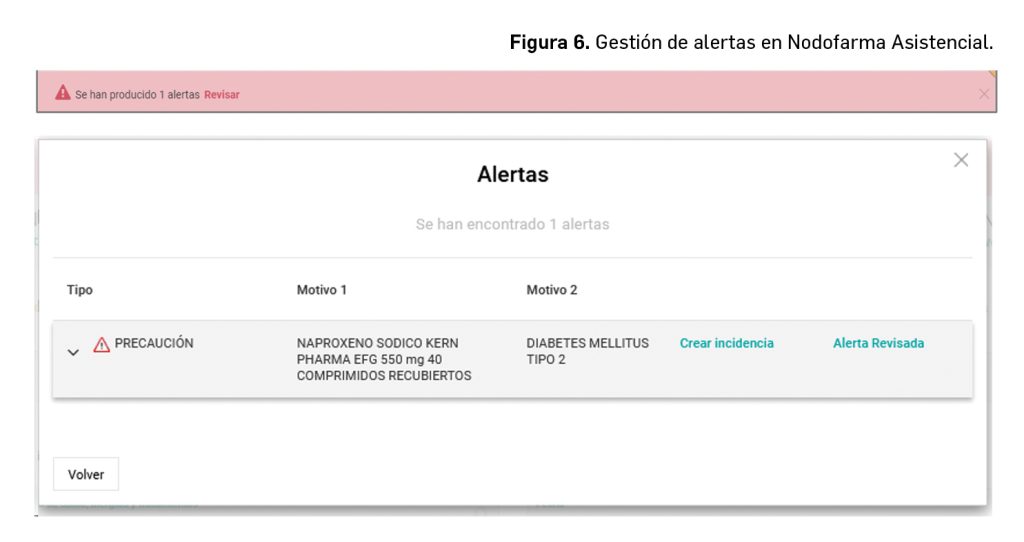
Además, Nodofarma Asistencial incorpora toda la información contenida en la base de datos del medicamento, Bot PLUS, del Consejo General. Gracias a esto facilita toda la información contenida en las fichas técnicas de los medicamentos e información sobre problemas de salud. Con ello se dispone de un sistema de alertas que avisa al farmacéutico de la información a tener en cuenta entre los tratamientos y las patologías del paciente (Figura 6).
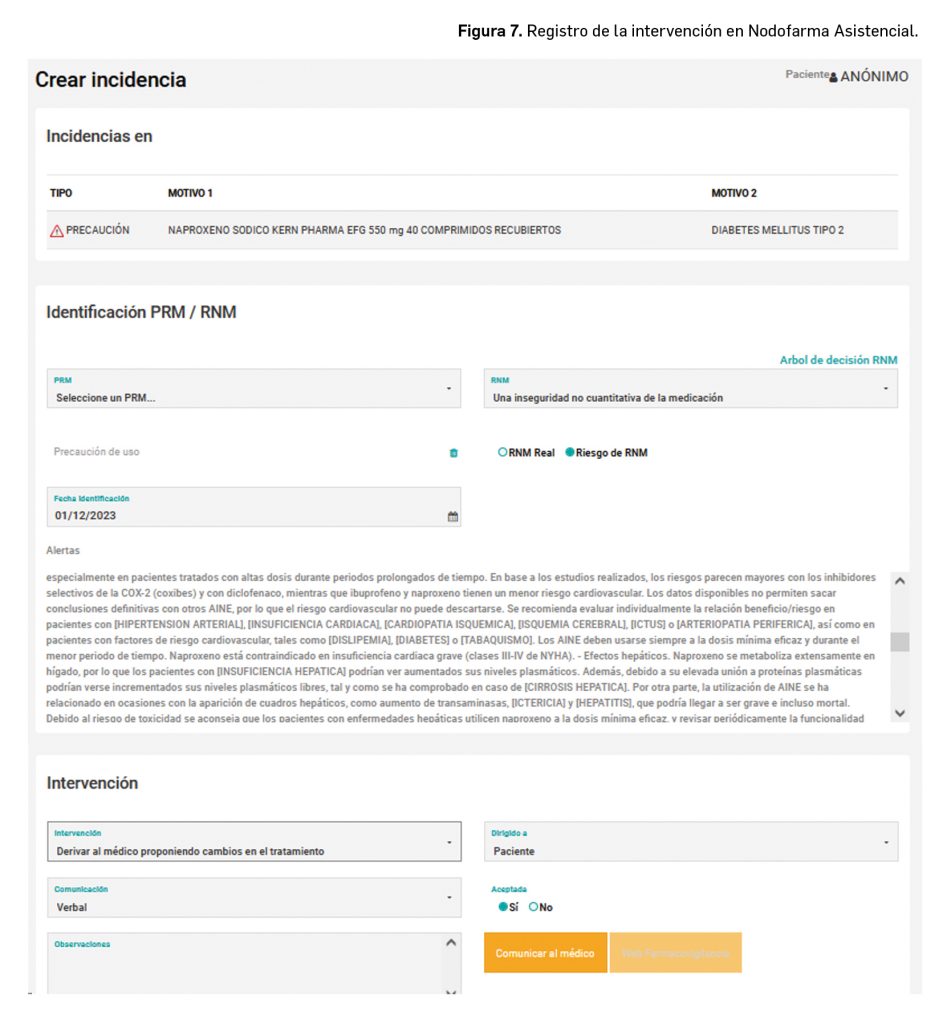
Estas alertas son: reacciones adversas, interacciones (entre fármaco-fármaco y/o fármaco-alimento), contraindicaciones, duplicidades y precauciones. Además, se ordenan de mayor a menor importancia y gravedad, para facilitar su visualización y estudio por el farmacéutico y, con ello, prevenir situaciones que pueden resultar incluso fatídicas (Figura 7).
Finalmente se finalizará el Servicio con la entrega o no del medicamento. Si no se detectan incidencias, o se detectan pero se pueden resolver en el momento, se realizará la entrega del medicamento con Información Personalizada del Medicamento (IPM) en caso de una dispensación de inicio o con educación sanitaria, en caso de una dispensación de continuación. En conclusión, hay que destacar que mediante la utilización de una herramienta de registro de Servicios Asistenciales como es Nodofarma Asistencial el farmacéutico comunitario puede registrar y documentar la actividad profesional realizada, la identificación de incidencias así como las intervenciones y actuaciones realizadas. Además, cuando haya identificado una incidencia y sea posible, podrá registrar el resultado obtenido en la salud del paciente (mejora, empeora, sigue igual) con el objetivo de evaluar del Servicio. Todo ello en aras de facilitar su labor asistencial.
La lipoproteína (a) o Lp(a) es una lipoproteína de síntesis hepática, estructuralmente similar a las lipoproteínas de baja densidad o LDL. La Lp(a) está en parte compuesta por la apolipoproteína (a) o Apo(a), que se acumula en las lesiones ateroscleróticas favoreciendo la trombogénesis. Aunque las funciones de la Lp(a) no se conocen completamente, se sabe que sus niveles sanguíneos correlacionan bien1 con el riesgo cardiovascular –por ejemplo, con el riesgo de infarto–. Debido a ello existe interés por desarrollar nuevos fármacos que permitan reducir los niveles de Lp(a), lo cual hipotéticamente debería redundar en una reducción del riesgo cardiovascular. En este sentido, recientemente se han presentado los resultados de dos estudios independientes con dos nuevos fármacos en investigación que actúan mediante este enfoque: lepodisirán, un ARN pequeño de interferencia, y muvalaplin, una molécula pequeña que actúa como inhibidor de la formación de la Lp(a).
En el caso de lepodisirán, se diseñó un estudio de fase 1 de escalada de dosis en el que los participantes, sin enfermedad cardiovascular pero con niveles elevados de Lp(a) (≥ 30 mg/dl), fueron aleatorizados para recibir placebo (n= 12) o una dosis única subcutánea de 4, 12, 32, 96, 304 o 608 mg del fármaco (n= 6 para cada grupo de dosis). Tras un seguimiento máximo de 48 semanas, se observó una reducción dosis-dependiente de los niveles de Lp(a) respecto al nivel basal (placebo: – 5%; lepodisirán 4 mg: – 41%; 12 mg: – 59%; 32 mg: – 76%; 96 mg: – 90%; 304 mg: – 96%; 608 mg: – 97%). El tratamiento fue bien tolerado, observándose un único evento adverso grave, no relacionado con el tratamiento.
Muvalaplin, por su parte, es un fármaco de administración oral cuyos efectos sobre el nivel de Lp(a) se han investigado en un estudio de fase 1, aleatorizado, doble ciego y de escalada de dosis que contó con un diseño de grupos paralelos, en el que 55 participantes –con independencia de su nivel basal de Lp(a)– fueron asignados a un grupo de dosis única vs. placebo y 59 –con niveles de Lp(a) ≥ 30 mg/dl– fueron asignados a un grupo de dosis múltiple vs. placebo (una dosis al día). Se observó una reducción de los niveles de Lp(a) a las 24 horas tras la administración de la primera dosis, con una reducción máxima frente a placebo del 65% a los 14 días (fin de seguimiento) con dosis repetidas superiores a 100 mg/día. En este periodo, no se observaron efectos adversos de relevancia clínica.
Los resultados obtenidos en estos ensayos clínicos abren la puerta a evaluar la relación entre la reducción de los niveles de Lp(a) con la reducción del riesgo cardiovascular pues, aunque la correlación entre ambas variables está bien descrita a nivel poblacional, el efecto de la reducción del nivel de Lp(a) por medios farmacológicos todavía no se conoce. No obstante, se trata de una opción interesante que podría abrir una nueva vía de tratamiento para una condición de elevada prevalencia a nivel mundial.
Los virus oncolíticos son virus modificados genéticamente con capacidad para infectar a todas las células pero que solo se replican en las células tumorales, acumulándose en gran cantidad dentro de éstas y liberándose de manera masiva, provocando la muerte selectiva de la célula tumoral. Talimogén laherparepvec (T-VEC), autorizado en 2015 en la UE, es una variante del virus del Herpes simplex de tipo 1 (HSV-1) genéticamente modificada para replicarse selectivamente dentro de los tumores y producir, dentro de estos, el factor estimulante de colonias de granulocitos y monocitos (GM-CSF; Granulocyte Macrophage Colony-Stimulating Factor), provocando la muerte de las células tumorales y la liberación de antígenos tumorales, lo que a su vez promueve una respuesta antitumoral inmune sistémica y una respuesta de células T efectoras.
La autorización de T-VEC en la UE se basó en un estudio en pacientes con melanoma metastásico, en el cual un 16,3% de los pacientes con tumor irresecable mostraron respuesta sostenida frente al 2,1% de los tratados con GM-CSF (p < 0,001). En este estudio de fase 3, la monoterapia con T-VEC se asoció con una mayor probabilidad de supervivencia libre de recaída (SLR) en combinación con cirugía vs. cirugía sola a los 2 años (29,5% vs. 16,9%; HR: 0,75) en pacientes con melanoma resecable, con un efecto persistente al tercer año (28,1% vs. 16,9%; HR: 0,74). Los resultados tras un seguimiento de 5 años (mediana de 63,3 meses) continúan indicando un beneficio sostenido, con una SLR del 22,3% con T-VEC + cirugía vs. 15,3% con cirugía sola (HR: 0,76) y una supervivencia global también superior para T-VEC + cirugía (77,3% vs. 62,7%; HR: 0,54). El perfil de seguridad del fármaco se considera aceptable, especialmente teniendo en cuenta la gravedad de la enfermedad a tratar. Los eventos adversos más comunes son enfermedad gripal, pirexia, fatiga, escalofríos y dolor de cabeza. Entre los graves, ningún evento presentó una incidencia > 3%, siendo el más frecuente la celulitis.
Por tanto, T-VEC se confirma como una opción efectiva a largo plazo en el tratamiento de pacientes con melanoma avanzado. El tratamiento, aunque autorizado en la Unión Europea, todavía no se encuentra comercializado en España.
El trasplante de precursores hematopoyéticos (TPH) es una opción terapéutica ampliamente estudiada y que ha sido empleada principalmente –aunque no de manera exclusiva– en el tratamiento de determinadas neoplasias hematológicas, en las cuales el TPH puede representar la única opción curativa. Sin embargo, sus aplicaciones no se limitan a alteraciones de tipo neoplásico ni hematológico y, de hecho, la utilidad de esta intervención ha sido investigada en muchas otras enfermedades, por ejemplo en enfermedades autoinmunes como la esclerosis múltiple (EM). De hecho, en estudios clínicos aleatorizados en pacientes con EM se ha podido documentar la remisión prolongada de la progresión de la enfermedad en pacientes que no respondían al tratamiento habitual. A pesar de su eficacia, el TPH se asocia con una elevada toxicidad y efectos adversos que lo descartan como opción en muchos pacientes.
En un nuevo estudio, un equipo de investigadores ha llevado a cabo un trasplante con precursores no hematopoyéticos en pacientes con EM progresiva, para la cual no existe hasta ahora un tratamiento efectivo. Concretamente, utilizaron células madre neurales, que presentan determinadas ventajas frente a los progenitores hematopoyéticos, como un bajo riesgo tumorigénico y baja capacidad de proliferación.
El procedimiento consistió en la administración intracerebroventricular de células madre neurales alogénicas procedentes de un único donante –un feto humano resultado de un aborto espontáneo– a 15 pacientes con EM secundaria progresiva (EMSP)1, en el marco de un estudio de fase 1, abierto, multicéntrico y de escalada de dosis. Tras un seguimiento de 12 meses, no se observaron muertes ni eventos adversos graves y durante este periodo los pacientes mostraron signos de estabilización de la enfermedad, sin recidivas ni síntomas de progresión.
A pesar de los esperanzadores resultados obtenidos en este estudio, debe tenerse en cuenta que se trata de un estudio de fase 1 con una muestra reducida y un periodo de seguimiento todavía corto. Además, la necesidad de administrar un tratamiento concomitante de tipo antiinflamatorio e inmunosupresor para limitar la capacidad inmunogénica del trasplante alogénico puede constituir un factor de confusión. Estos aspectos deberán valorarse en estudios clínicos más amplios, aunque por ahora abren la puerta a una interesante nueva vía terapéutica.
La hipertensión arterial pulmonar se caracteriza por un incremento progresivo de la resistencia vascular periférica, que puede conducir a un fallo ventricular derecho y a la muerte. Los mecanismos patogénicos de la enfermedad incluyen una disfunción endotelial pulmonar, lo cual provoca una deficiencia en la producción de vasodilatadores, como el óxido nítrico y prostaciclina, y una sobreexpresión de vasoconstrictores, como endotelina-1. Por ahora no se dispone de un tratamiento curativo, por lo que los fármacos empleados se dirigen a la reducción de los síntomas.
El desarrollo de sotatercept, una proteína de fusión compuesta por el dominio extracelular del receptor IIA de la activina, plantea una nueva aproximación al tratamiento de esta enfermedad. El fármaco en investigación actúa inhibiendo la señaFlización producida por la activina, lo que se relaciona con una menor proliferación de células vasculares y revierte el remodelado vascular y del ventrículo derecho.
Recientemente se han publicado los resultados de un estudio de fase 3, doble ciego, en el que se aleatorizó a 323 pacientes en proporción 1:1 para recibir sotatercept o placebo cada 3 semanas. La variable primaria de eficacia del estudio fue el cambio respecto a la línea de base en la prueba de la marcha de 6 minutos1 en la semana 24, con una diferencia estimada de 40,8 metros favorable a sotatercept (IC95%: 27,5 – 54,1; p < 0,001). Estos resultados positivos para el fármaco en investigación se vieron apoyados por los resultados en variables secundarias, como la resistencia pulmonar vascular, los niveles de NT-proBNP y cambios en la exploración hemodinámica. Además, tras un seguimiento mediano de 32,7 semanas, sotatercept redujo el riesgo de progresión o muerte respecto a placebo (HR: 0,16; IC95%: 0,08 – 0,35). Los resultados tras un año de seguimiento en un estudio abierto de extensión son indicativos del mantenimiento de la eficacia. En cuanto a los resultados de seguridad, la frecuencia de eventos adversos fue solo ligeramente superior en los pacientes que recibieron sotatercept (87,5% vs. 84,7%), siendo los más comunes dolor de cabeza, COVID-19, náuseas, diarrea, fatiga y sangrado de la nariz.
La hipertensión pulmonar arterial es una enfermedad progresiva y que compromete la vida de los pacientes, lo que resalta la importancia de desarrollar nuevos fármacos capaces de frenar e incluso revertir el remodelado, que se asocia en última instancia con un empeoramiento de la enfermedad y mayor riesgo de muerte. Su incorporación dependerá de la evaluación de la eficacia y seguridad que realicen las Agencias reguladoras –la EMA en el caso de la Unión Europea–, pero los datos presentados resultan esperanzadores.
Eficacia: el logro de los objetivos propuestos (sin considerar costes)
Eficiencia: el logro de los objetivos haciendo uso del mínimo de recursos necesarios (coste)
Eficiencia económica: una intervención es eficiente económicamente cuando los beneficios sociales que reporta son mayores que su coste.
Eficiencia técnica: capacidad de una unidad económica (por ejemplo, hospital) para producir el máximo posible, dado un conjunto de inputs (tecnología, trabajo y capital). Es decir, no puede producir más sin incrementar la cantidad de inputs.
Eficiencia asignativa: capacidad de una unidad económica (por ejemplo, hospital) para escoger el conjunto óptimo de inputs, dados los correspondientes precios. Se trata de producir lo máximo a partir de una combinación de inputs que con el mínimo coste permitirá alcanzar un output determinado a unos precios establecidos.
Equidad: se basa en la igualdad (según juicios de valor) como valor social fundamental
Equidad vertical: tratamiento desigual para los que son desiguales (quién ha de pagar; progresividad en la financiación).
Equidad horizontal: igual tratamiento para iguales (necesidad).
DE LAS POLÍTICAS DE SALUD A LA GESTIÓN SANITARIA
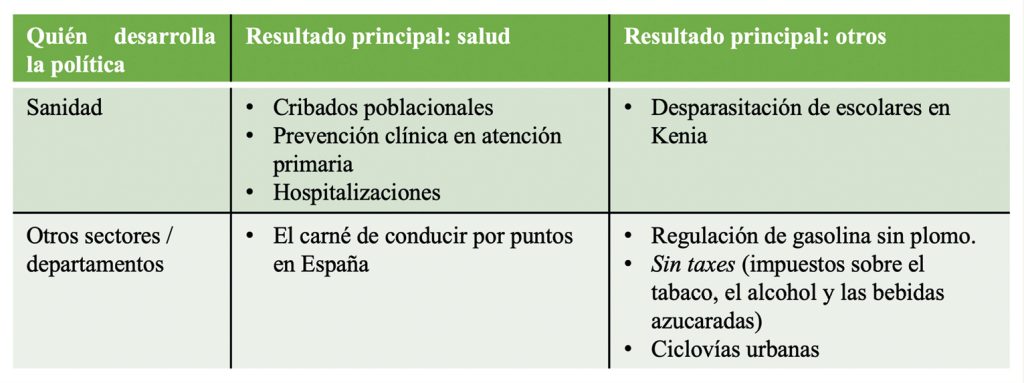
Las políticas de salud actúan sobre los servicios de salud, los estilos de vida y el entorno. Algunos ejemplos se muestran en la tabla a continuación, teniendo en cuenta los inputs y outputs principales de algunas políticas sanitarias y no sanitarias concretas.
NIVELES DE GESTIÓN SANITARIA
Macrogestión o política sanitaria: intervención del Estado para aumentar la equidad y corregir disfunciones del mercado (externalidades, regulación de monopolios y corrección de la información imperfecta).
Corresponden las labores de regulación, planificación, financiación y organización sanitarias, encaje institucional, descentralización y buen gobierno.
Mesogestión o gestión de centros: busca coordinar y motivar en un entorno muy regulado con apoyo en las ciencias de gestión.
Funciones: planificación, dirección, organización, coordinación y control.
Microgestión o gestión clínica: proceso de toma de decisiones en la práctica clínica orientado a conseguir el máximo beneficio para el paciente, mejorando la calidad del servicio sanitario
Tres elementos diferenciadores: perspectivas de los agentes, prioridades y responsabilidades.